
Publications
| No. | Publication Details | Abstract Image |
|---|---|---|
259 | Synthesis, Thermochemistry, and Cure Behavior of Oligocyclobutane-Containing Prepolymers Relevant to Propellant ApplicationsKaitlin Nachtrieb, Cherish Nie, Paul J. Chirik, and Megan Mohadjer Beromi ACS Appl. Polym. Mater. 2024, 6, 9, 5171–5182 AbstractWhile hydroxy-terminated polybutadiene (HTPB) has been traditionally employed as a binder prepolymer for solid rocket booster applications, the hydrocarbon main chain is energetically inert and contributes little to the overall energy content of the formulation. Here, (1,n′-divinyl)oligocyclobutane (DVOCB), a structural isomer of polybutadiene containing 1,3-enchained cyclobutanes in the polymer backbone, is investigated as a prepolymer candidate. The synthetic modularity of DVOCB is also demonstrated through postsynthetic dihydroxylation to generate hydroxy-terminated oligocyclobutane (HTOC), the direct structural isomer of HTPB. Because the main chain of these cyclobutyl-containing prepolymer candidates is composed of strained four-membered rings as repeat units, the use of these molecules in binder applications was targeted to increase the energy density of the formulations. The measurement of the energetic content of both oligomers by constant volume (bomb) calorimetry indicated that DVOCB and HTOC are 16−18% more energetic than their linear congeners. Further, their ability to participate as synthetic analogues of HTPB was demonstrated through thermal, rheological, and curing studies to generate polyurethanes or polythioethers. | 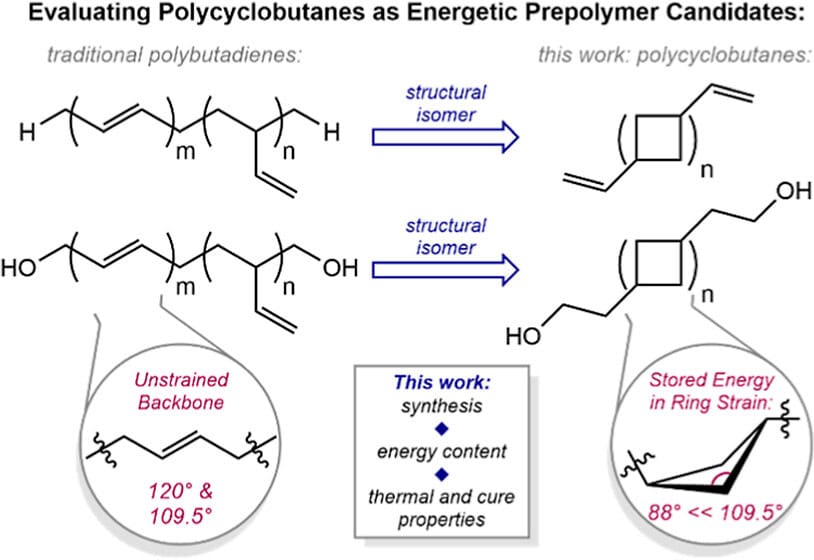 |
258 | C(sp3)–C(sp3) Reductive Elimination from (Phenoxyimine)Cobalt(III)(CH3)2(PMe3)2 ComplexesL. Reginald Mills, Junho Kim, Eric M. Simmons, Steven R. Wisniewski, and Paul J. Chirik Organometallics 2024, 43, 9, 1021–1029 AbstractA series of six-coordinate, idealized octahedral phenoxyimine (FI)–cobalt(III) dimethyl bis(trimethylphosphine) complexes was synthesized and characterized by NMR spectroscopy and single-crystal X-ray diffraction. The thermal stability of the parent Ph-FI complex was evaluated at 60 °C in benzene-d6, and a 13(1):1 ratio of ethane to methane was observed. The major detectable cobalt product of this reaction was the bis(chelate) cobalt derivative (Ph-FI)2Co formed by disproportionation of the (FI)cobalt(I) product following ethane reductive elimination. Addition of excess PMe3 inhibited C(sp3)–C(sp3) reductive elimination, consistent with phosphine dissociation preceding C–C bond formation from a five-coordinate (FI)cobalt(III) dimethyl intermediate. The reductive elimination of substituted (R-FI)cobalt(III) dimethyl bis(trimethylphosphine) compounds was evaluated in acetonitrile-d3, where ligands bearing electron-donating aniline substituents underwent reductive elimination the fastest and electron-withdrawing substituents the slowest. These data support a buildup of positive charge in the rate-limiting step, consistent with the formation of a more electropositive five-coordinate cobalt center prior to rate-limiting C–C reductive elimination. Attempted synthesis of a cobalt(III) dimethyl complex bearing a sterically hindered FI ligand with a tert-butyl substituent ortho to the phenol led exclusively to the corresponding bis(chelate) cobalt derivative, whose formation was rationalized by steric destabilization of pre–reductive elimination intermediates. | 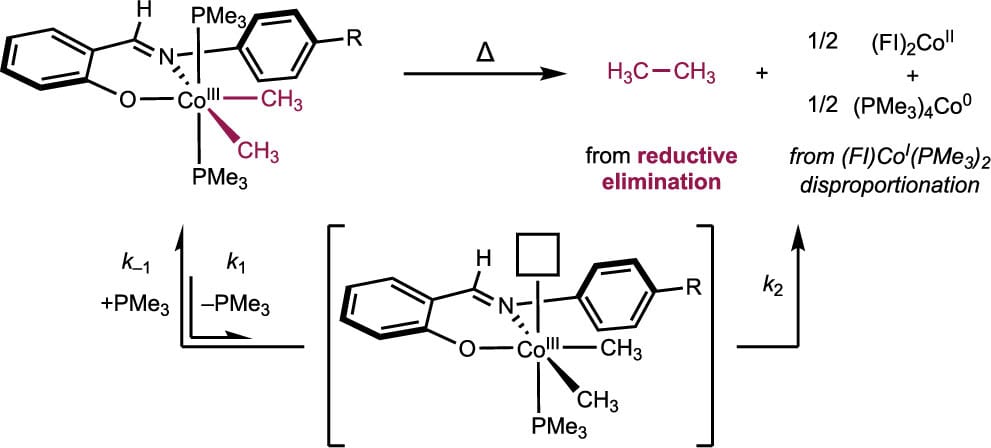 |
257 | (Phenoxyimine)nickel-Catalyzed C(sp2)–C(sp3) Suzuki–Miyaura Cross-Coupling: Evidence for a Recovering Radical Chain MechanismL. Reginald Mills, Eric M. Simmons, Heejun Lee, Eva Nester, Junho Kim, Steven R. Wisniewski, Matthew V. Pecoraro, and Paul J. Chirik J. Am. Chem. Soc. 2024, 146, 14, 10124–10141 AbstractPhenoxyimine (FI)–nickel(II)(2-tolyl)(DMAP) compounds were synthesized and evaluated as precatalysts for the C(sp2)–C(sp3) Suzuki–Miyaura cross coupling of (hetero)arylboronic acids with alkyl bromides. With 5 mol % of the optimal (MeOMeFI)Ni(Aryl)(DMAP) precatalyst, the scope of the cross-coupling reaction was established and included a variety of (hetero)arylboronic acids and alkyl bromides (>50 examples, 33–97% yield). A β-hydride elimination–reductive elimination sequence from reaction with potassium isopropoxide base, yielding a potassium (FI)nickel(0)ate, was identified as a catalyst activation pathway that is responsible for halogen atom abstraction from the alkyl bromide. A combination of NMR and EPR spectroscopies identified (FI)nickel(II)–aryl complexes as the resting state during catalysis with no evidence for long-lived organic radical or odd-electron nickel intermediates. These data establish that the radical chain is short-lived and undergoes facile termination and also support a “recovering radical chain” process whereby the (FI)nickel(II)–aryl compound continually (re)initiates the radical chain. Kinetic studies established that the rate of C(sp2)–C(sp3) product formation was proportional to the concentration of the (FI)nickel(II)–aryl resting state that captures the alkyl radical for chain propagation. The proposed mechanism involves two key and concurrently operating catalytic cycles; the first involving a nickel(I/II/III) radical propagation cycle consisting of radical capture at (FI)nickel(II)–aryl, C(sp2)–C(sp3) reductive elimination, bromine atom abstraction from C(sp3)–Br, and transmetalation; and the second involving an off-cycle catalyst recovery process by slow (FI)nickel(II)–aryl → (FI)nickel(0)ate conversion for nickel(I) regeneration. | 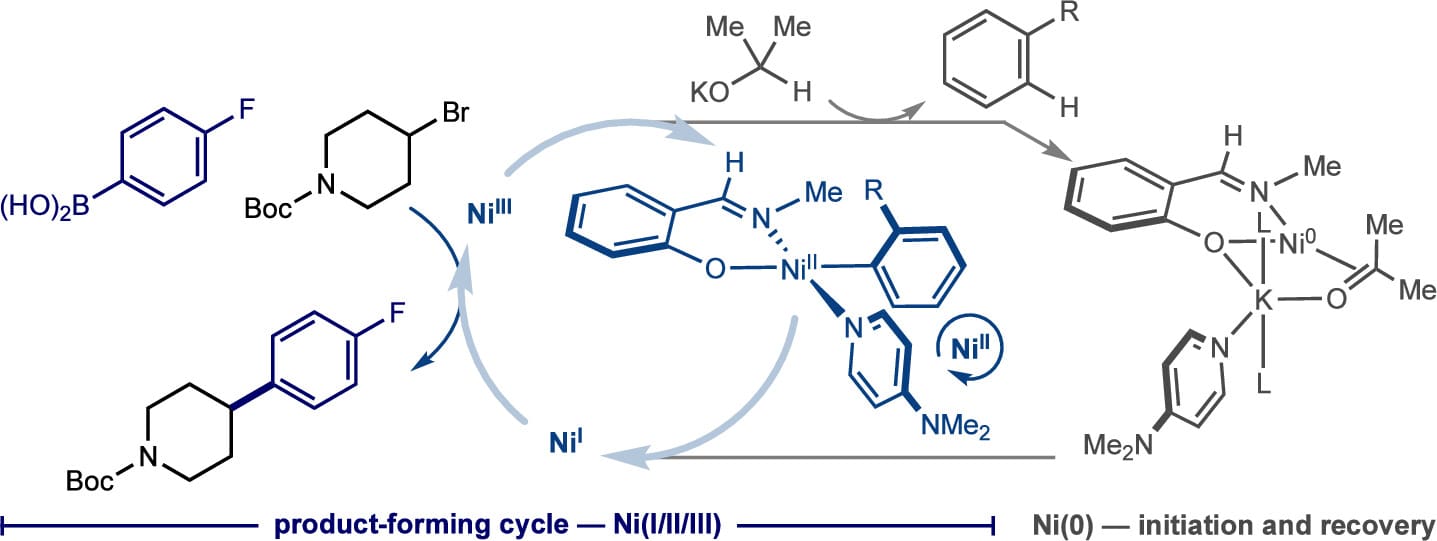 |
256 | Ligand Field Sensitive Spin Acceleration in the Iron-Catalyzed [2 + 2] Cycloaddition of Unactivated Alkenes and DienesHanna H. Cramer, Coralie Duchemin, Carli B. Kovel, Junho Kim, Matthew V. Pecoraro, and Paul J. Chirik J. Am. Chem. Soc. 2024, 146, 14, 9947–9956 AbstractRedox-active pyridine(diimine) (PDI) iron catalysts promote the reversible [2 + 2] cycloaddition of alkenes and dienes to cyclobutane derivatives that have applications ranging from fuels to chemically recyclable polymers. Metallacycles were identified as key intermediates, and spin crossover from the singlet to the triplet surface was calculated to facilitate the reductive coupling step responsible for the formation of the four-membered ring. In this work, a series of sterically and electronically differentiated PDI ligands was studied for the [2 + 2] cycloaddition of ethylene and butadiene to vinylcyclobutane. Kinetic studies revealed that the fastest and slowest turnover were observed with equally electron-deficient supporting ligands that either feature phenyl-substituted imine carbon atoms (MeBPDI) or a pyrazine core (MePZDI). While the oxidative cyclization was comparatively slow for both catalysts, the rate of reductive coupling─determined by stoichiometric 13C2H4 labeling studies─correlated with the turnover frequencies. Two-state density functional theory studies and the distinct electronic structures of related (iPrBPDI) and (iPrPZDI) iron methyl complexes revealed significantly different ligand field strengths due to either diminished ligand σ-donation (MeBPDI) or promoted metal π-backbonding (MePZDI). Spin acceleration, leading to fast reductive coupling and catalytic turnover, was promoted in the case of the weaker ligand field and depends on both the nature and position of the electron-withdrawing group. This study provides strong evidence for the role of two-state reactivity in C(sp3)–C(sp3) bond formation and insights on how ligand design either promotes or inhibits spin acceleration in earth-abundant metal catalysis. | ![Ligand Field Sensitive Spin Acceleration in the Iron-Catalyzed [2 + 2] Cycloaddition of Unactivated Alkenes and Dienes](/wp-content/uploads/2024/04/img-publ-256.jpeg) |
255 | A butadiene-derived semicrystalline polyolefin with two-tiered chemical recyclabilityCherish Nie, Shawn M. Maguire, Callie W. Zheng, Megan Mohadjer Beromi, Richard A. Register, Rodney D. Priestley, Emily C. Davidson, Paul J. Chirik AbstractCommodity polyolefins account for the majority of plastic waste but are challenging to depolymerize or upcycle due to the high thermal and chemical stability of hydrocarbon polymer backbones. Here, we report a class of polyolefin that undergoes clean and selective chemical recycling at both the oligomeric and polymeric stages. Iron-catalyzed [2+2] cycloaddition of the monomer butadiene formed telechelic oligomers (1,n′-divinyl)-oligocyclobutane (DVOCB) that undergo deoligomerization back to butadiene. From the oligomer DVOCB, chain extension using acyclic diene metathesis (ADMET) yielded the polyolefin pDVOCB. Reverse ADMET of pDVOCB enabled full recovery of the telechelic oligomer. pDVOCB polymers are semicrystalline hydrocarbon polymers with high melting temperatures (Tm > 230°C) that have excellent chemical and hydrolytic stability and comparable mechanical properties to commodity polyolefins, such as polypropylene and polyethylene. Thus, this work demonstrates a class of semicrystalline polyolefins that offers two tiers of chemical recycling. | 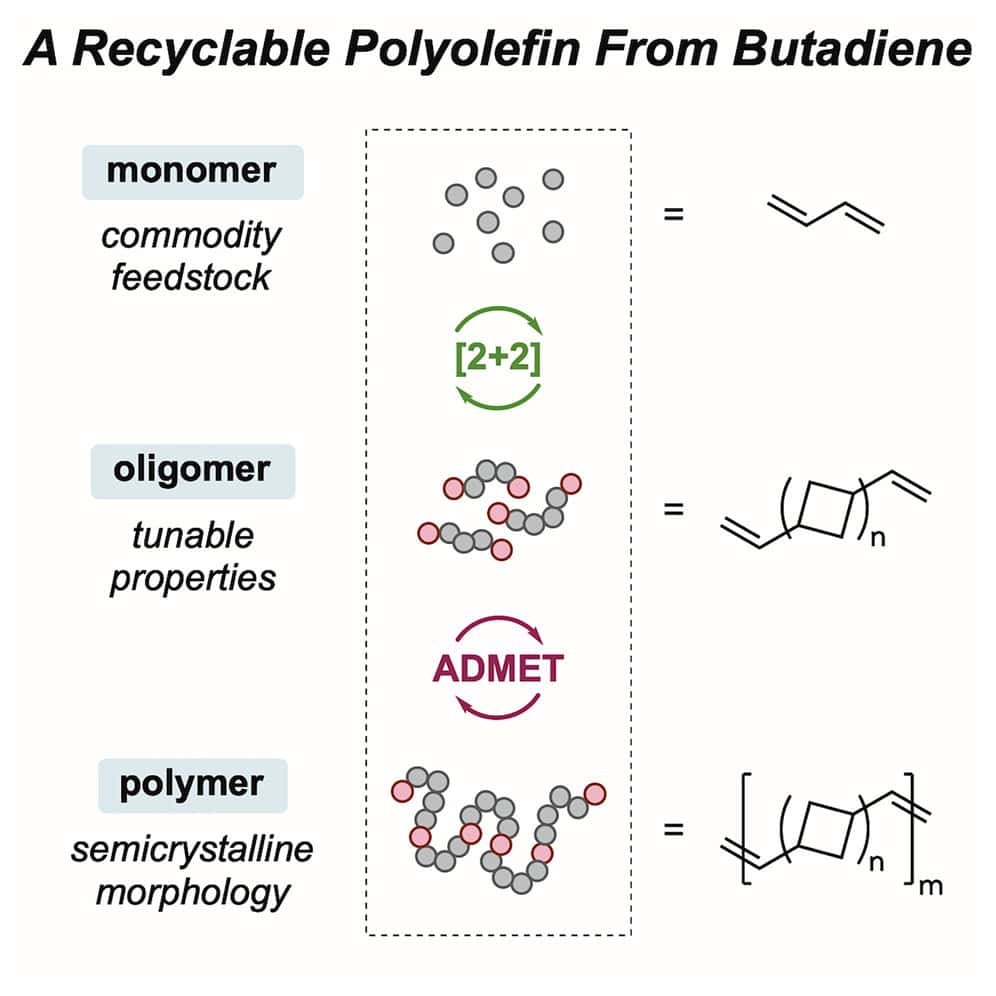 |
254 | Kinetic and thermodynamic control of C(sp2)–H activation enables site-selective borylationJose B. Roque, Alex M. Shimozono, Tyler P. Pabst, Gabriele Hierlmeier, Paul O. Peterson, and Paul J. Chirik AbstractCatalysts that distinguish between electronically distinct carbon-hydrogen (C–H) bonds without relying on steric effects or directing groups are challenging to design. In this work, cobalt precatalysts supported by N-alkyl-imidazole–substituted pyridine dicarbene (ACNC) pincer ligands are described that enable undirected, remote borylation of fluoroaromatics and expansion of scope to include electron-rich arenes, pyridines, and tri- and difluoromethoxylated arenes, thereby addressing one of the major limitations of first-row transition metal C–H functionalization catalysts. Mechanistic studies established a kinetic preference for C–H bond activation at the meta-position despite cobalt-aryl complexes resulting from ortho C–H activation being thermodynamically preferred. Switchable site selectivity in C–H borylation as a function of the boron reagent was thereby preliminarily demonstrated using a single precatalyst. | 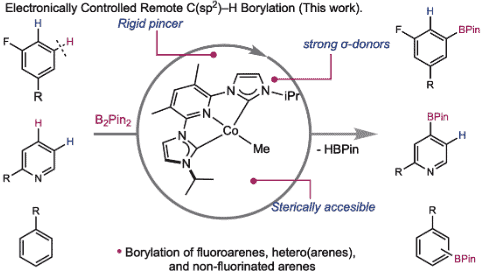 |
253 | Iridium-Catalyzed Hydrogenation of a Phenoxy Radical to the Phenol: Overcoming Catalyst Deactivation with Visible Light IrradiationJunho Kim, Yoonsu Park, and Paul J. Chirik Inorg. Chem. 2023, 62, 48, 19582–19592 AbstractPiano-stool iridium hydride complexes bearing phenylpyridine ligands are effective precatalysts for promoting the formation of element-hydrogen bonds using H2 as the stoichiometric H-atom source. Irradiation with blue light resulted in a profound enhancement of catalyst turnover for the iridium-catalyzed hydrogenation of the aryloxyl radical 2,4,6-tBu3-C6H2O• to the corresponding phenol. Monitoring the progress of the reaction revealed the formation of an iridium 3,3-dimethyl-2,3-dihydrobenzofuranyl compound arising from two C–H activation events following the proton-coupled electron transfer (PCET) step. Under thermal conditions, this compound was inactive for catalytic aryloxide hydrogenation, representing a deactivation pathway. Irradiation with blue light under H2 released the free heterocycle and regenerated the piano-stool iridium hydride precatalyst, establishing a pathway for catalyst recovery and overall enhanced turnover. | 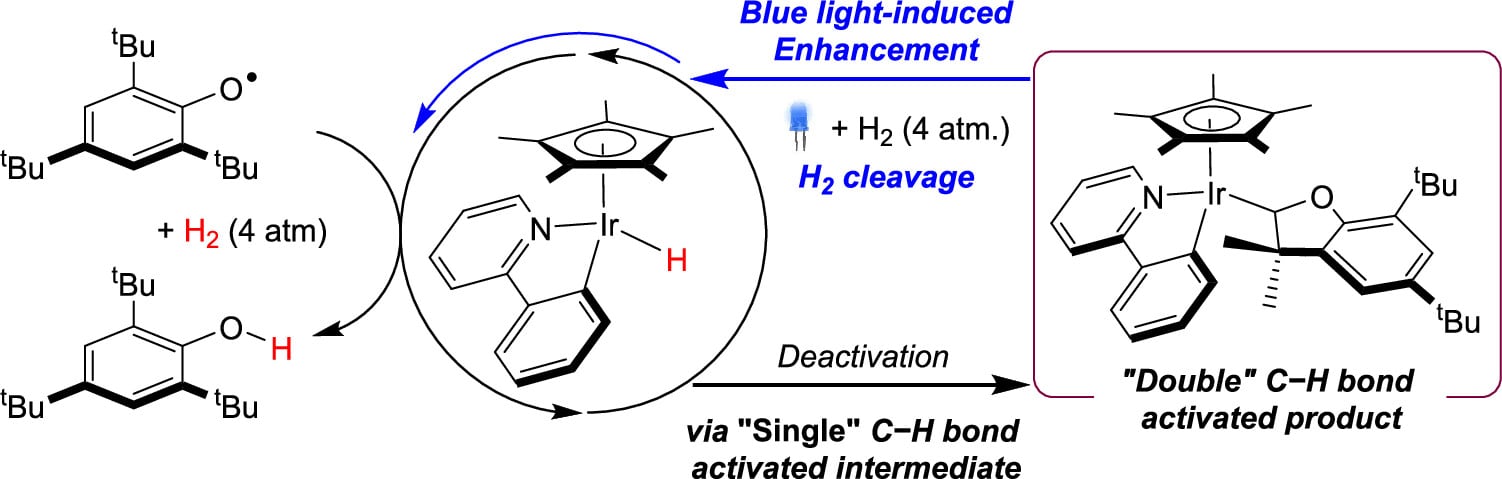 |
252 | Phenoxythiazoline (FTz)-Cobalt(II) Precatalysts Enable C(sp2)–C(sp3) Bond-Formation for Key Intermediates in the Synthesis of Toll-like Receptor 7/8 AntagonistsDr. L. Reginald Mills, Francesca Di Mare, Dr. David Gygi, Dr. Heejun Lee, Dr. Eric M. Simmons, Junho Kim, Dr. Steven R. Wisniewski, Prof. Dr. Paul J. Chirik Angew. Chem. Int. Ed. 2023, e202313848 AbstractEvaluation of the relative rates of the cobalt-catalyzed C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling between the neopentylglycol ester of 4-fluorophenylboronic acid and N-Boc-4-bromopiperidine established that smaller N-alkyl substituents on the phenoxyimine (FI) supporting ligand accelerated the overall rate of the reaction. This trend inspired the design of optimal cobalt catalysts with phenoxyoxazoline (FOx) and phenoxythiazoline (FTz) ligands. An air-stable cobalt(II) precatalyst, (FTz)CoBr(py)3 was synthesized and applied to the cross-coupling of an indole-5-boronic ester nucleophile with a piperidine-4-bromide electrophile that is relevant to the synthesis of reported toll-like receptor (TLR) 7/8 antagonist molecules including afimetoran. Addition of excess KOMe⋅B(OiPr)3 improved catalyst lifetime due to attenuation of alkoxide basicity that otherwise resulted in demetallation of the FI chelate. A first-order dependence on the cobalt precatalyst and a saturation regime in nucleophile were observed, supporting turnover-limiting transmetalation and the origin of the observed trends in N-imine substitution. | 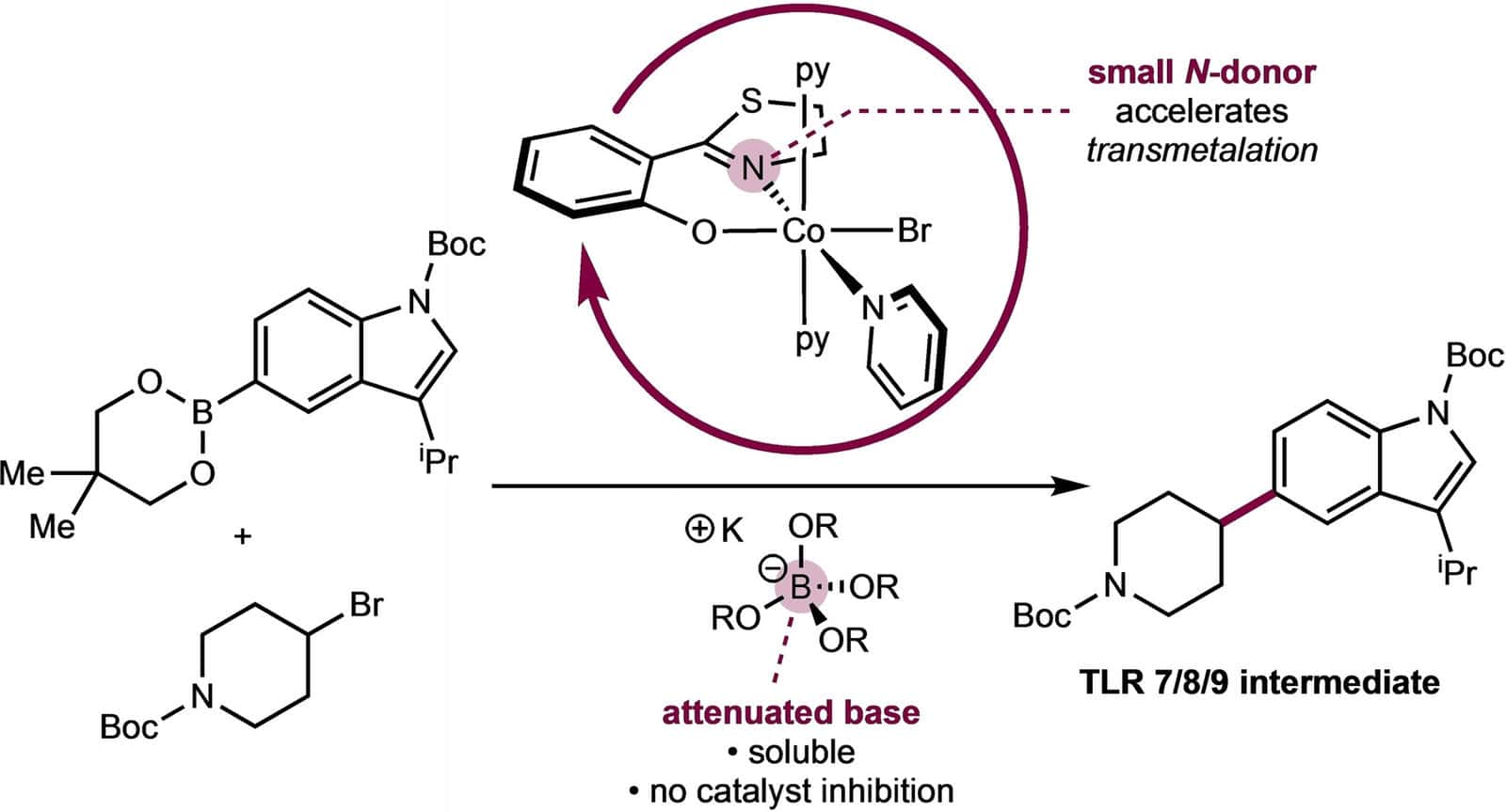 |
251 | Quinoline Pyridine(Imine) Iron Complexes as Catalysts for the 1,4-Hydrovinylation of 1,3-DienesCoralie Duchemin, Amelia I. Liu, Junho Kim, and Paul J. Chirik Organometallics 2023, 42, 21, 3109–3119 AbstractThe synthesis, characterization, and stoichiometric reactivity of a series of quinoline pyridine(imine) (R(R’)Q(R”)PI) (R = 2,6-Me, 2,6-iPr or 2,4,6-Cy; R’ = H or Cl, R″ = H or tBu) iron dichloride complexes is described. Treatment of (QPI)FeCl2 with two equivalents of methyl lithium furnished two examples of the corresponding (QPI)FeCH3 complexes. The molecular structures were established by X-ray diffraction, and the electronic structures were studied by magnetometry, 57Fe Mössbauer spectroscopy, and DFT calculations. The combined data support overall S = 3/2 ground states that are best described as high spin iron(II) complexes engaged in antiferromagnetic coupling with a chelate radical anion, similar to related pyridine(diimine) and terpyridine iron alkyl complexes. Bimolecular reductive elimination of ethane in the presence of butadiene afforded an iron crotyl complex bearing a cyclometalated QPI ligand, formed upon ligand-to-ligand hydrogen transfer (LLHT) or stepwise oxidative addition/reduction elimination from a putative (QPI) iron butadiene intermediate. The cyclometalated QPI iron crotyl complex proved to be a competent catalyst for the hydrovinylation of butadiene with ethylene and produced high selectivity for the hexa-1,4-diene isomer. Deuterium labeling experiments established H/D scrambling between ethylene, butadiene and the QPI ligand, consistent with background transfer hydrogenation reactivity and catalyst decomposition observed during catalysis. | 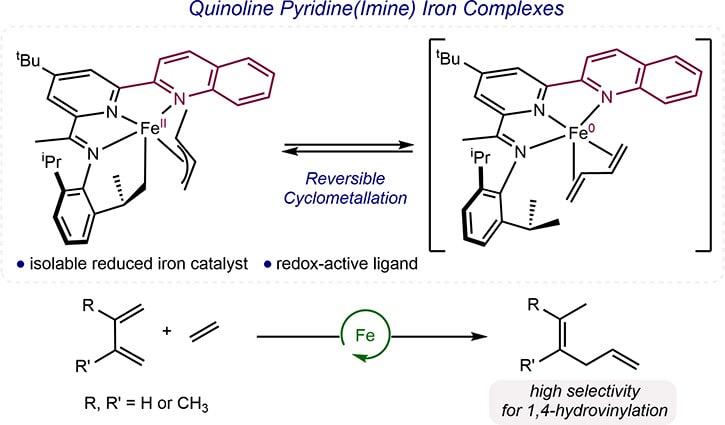 |
250 | Arene Insertion with Pincer-Supported Molybdenum-Hydrides: Determination of Site Selectivity, Relative Rates, and Arene Complex FormationGabriele Hierlmeier, Paolo Tosatti, Kurt Puentener, and Paul J. Chirik J. Am. Chem. Soc. 2023, 145, 38, 21027–21039 AbstractThe synthesis of phosphino(oxazoline)pyridine-supported molybdenum(0) cycloocta-1,5-diene complexes is described. Exposure of these complexes to dihydrogen in the presence of an arene resulted in insertion of the substrate into the molybdenum hydride bond and afforded the corresponding molybdenum cyclohexadienyl hydrides. For mono- and disubstituted arenes, the site selectivity for insertion of the most substituted bond increases with increasing size of the substituent from methyl to ethyl, iso-propyl, and tert-butyl. In contrast, 1,3,5-trisubstituted arenes underwent insertion with exclusive site selectivity. Relative rates of insertion were determined by competition experiments and established faster insertions for electron-rich arenes. Introduction of electron-withdrawing trifluoromethyl groups on the arene resulted in decreased relative rates of insertion and an increased rate for H2 reductive elimination, favoring formation of the corresponding molybdenum η6-arene complex. Studies on the reductive elimination of the cyclohexadienyl ligand with the hydride enabled the synthesis of an enantioenriched cyclohexa-1,3-diene. This study provides new insights into the ligand requirements for catalytic arene hydrogenation and a new strategy for selective arene reduction. | 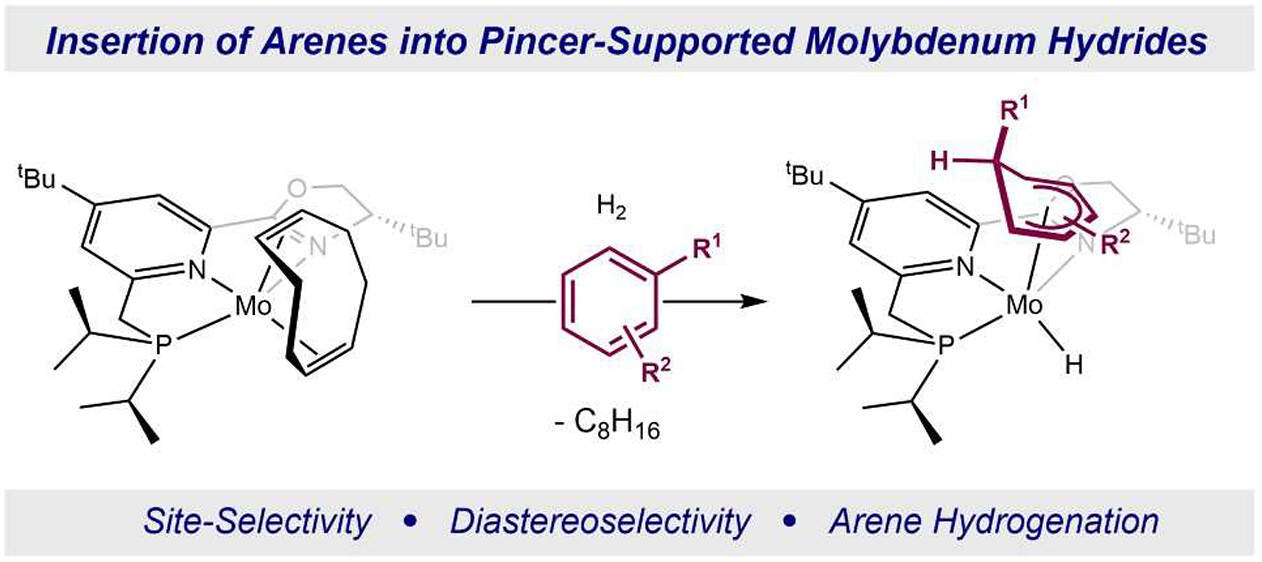 |
249 | Mechanistic Investigations of Phenoxyimine–Cobalt(II)-Catalyzed C(sp2)–C(sp3) Suzuki–Miyaura Cross-CouplingL. Reginald Mills, David Gygi, Eric M. Simmons, Steven R. Wisniewski, Junho Kim, and Paul J. Chirik J. Am. Chem. Soc. 2023, 145, 31, 17029–17041 AbstractThe mechanism of phenoxyimine (FI)−cobalt-catalyzed C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling was studied using a combination of kinetic measurements and catalytic and stoichiometric experiments. A series of dimeric (FI)cobalt(II) bromide complexes, [(4-CF3PhFI)CoBr]2, [(4-OMePhFI)CoBr]2, and [(2,6-diiPrPhFI)CoBr]2, were isolated and characterized by 1H and 19F NMR spectroscopies, solution and solid-state magnetic susceptibility, electron paramagnetic resonance (EPR) spectroscopy, X-ray crystallography, and diffusion-ordered NMR spectroscopy (DOSY). One complex, [(4-CF3PhFI)CoBr]2, was explored as a single-component precatalyst for C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling. Addition of potassium methoxide to [(4-CF3PhFI)CoBr]2 generated the corresponding (FI)cobalt(II) methoxide complex as determined by 1H and 19F NMR and EPR spectroscopies. These spectroscopic signatures were used to identify this compound as the resting state during catalytic C(sp2)–C(sp3) coupling. Variable time normalization analysis (VTNA) of in situ catalytic 19F NMR spectroscopic data was used to establish an experimental rate law that was first-order in a (FI)cobalt(II) precatalyst, zeroth-order in the alkyl halide, and first-order in an activated potassium methoxide–aryl boronate complex. These findings are consistent with turnover-limiting transmetalation that occurs prior to activation of the alkyl bromide electrophile. The involvement of boronate intermediates in transmetalation was corroborated by Hammett studies of electronically differentiated aryl boronic esters. Together, a cobalt(II)/cobalt(III) catalytic cycle was proposed that proceeds through a “boronate”-type mechanism. | 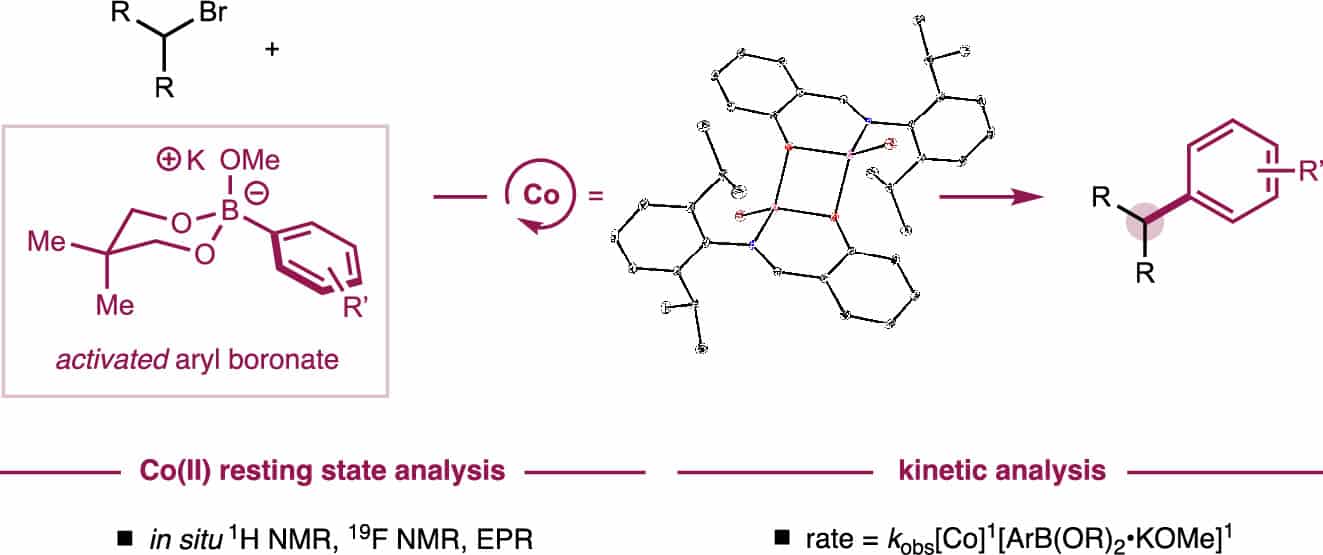 |
248 | CS-Symmetric Pyridine(diimine) Iron Methyl Complexes for Catalytic [2+2] Cycloaddition and Hydrovinylation: Metallacycle Geometry Determines SelectivityCoralie Duchemin, Junho Kim, and Paul J. Chirik AbstractA series of CS-symmetric (aryl,alkyl)-substituted pyridine(dimine) iron methyl (CyARPDI)FeCH3 complexes have been prepared, characterized, and evaluated as precatalysts for the [2+2]-cycloaddition of butadiene and ethylene. Mixtures of vinylcyclobutane and (Z)-hexa-1,4-diene were observed in each case. By comparison, C2v-symmetric, arylated (PDI) iron catalysts are exclusively selective for reversible [2+2]-cycloaddition to yield vinylcyclobutane. The alteration in the chemoselectivity of the catalytic reaction was investigated through a combination of precatalyst stability studies, identification of catalytic resting state(s), and 2H and 13C isotopic labeling experiments. While replacement of an aryl-imine substituent with an N-alkyl group decreases the stability of the formally iron(0) dinitrogen and butadiene complexes, two diamagnetic metallacycles were identified as catalyst resting states. Deuterium labeling and NOESY/EXSY NMR experiments support 1,4-hexadiene arising from catalytic hydrovinylation involving reversible oxidative cyclization leading to accessible cis-metallacycle. Cyclobutane formation proceeds by irreversible C(sp3)–C(sp3) bond-forming reductive elimination from a trans-metallacycle. These studies provide key mechanistic understanding into the high selectivity of bis(arylated) pyridine(diimine) iron catalysts for [2+2]-cycloaddition, unique, thus far, to this class of iron catalysts. | ![CS-Symmetric Pyridine(diimine) Iron Methyl Complexes for Catalytic [2+2] Cycloaddition and Hydrovinylation: Metallacycle Geometry Determines Selectivity](/wp-content/uploads/2023/07/img-publ-248.jpeg) |
247 | C(sp3)–C(sp2) Reductive Elimination versus β-Hydride Elimination from Cobalt(III) Intermediates in Catalytic C–H FunctionalizationWilliam G. Whitehurst, Junho Kim, Stefan G. Koenig, and Paul J. Chirik ACS Catal. 2023, 13, 13, 8700–8707 AbstractThe cationic bis(phosphine) cobalt(I) arene complex, [(depe)Co(η6-C7H8)][BAr4F] (depe = 1,2-bis(diethylphosphino)ethane; BAr4F = B[(3,5-(CF3)2)C6H3]4), was investigated as a precatalyst for three-component coupling of arenes, alkenes, and alkynes. Although a sterically attenuated catalyst was targeted with the goal of accelerating C–H functionalization, hydrovinylation derived from the alkene and alkyne was favored over three-component coupling. Remarkably, no catalytic hydrovinylation was observed in the absence of arene, demonstrating the role of C–H activation in the hydrovinylation process and ruling out a mechanism that involves β-H elimination from a cobalt(III) metallacycle. Deuterium labeling supported β-H elimination from a putative cobalt(III)–alkyl,aryl intermediate that forms after C–H activation by a cobalt(III) metallacycle. By varying the arene and alkene coupling partners, factors affecting the selectivity of C(sp3)–C(sp2) reductive elimination versus β-H elimination from the cobalt(III)–alkyl,aryl intermediate were determined. | 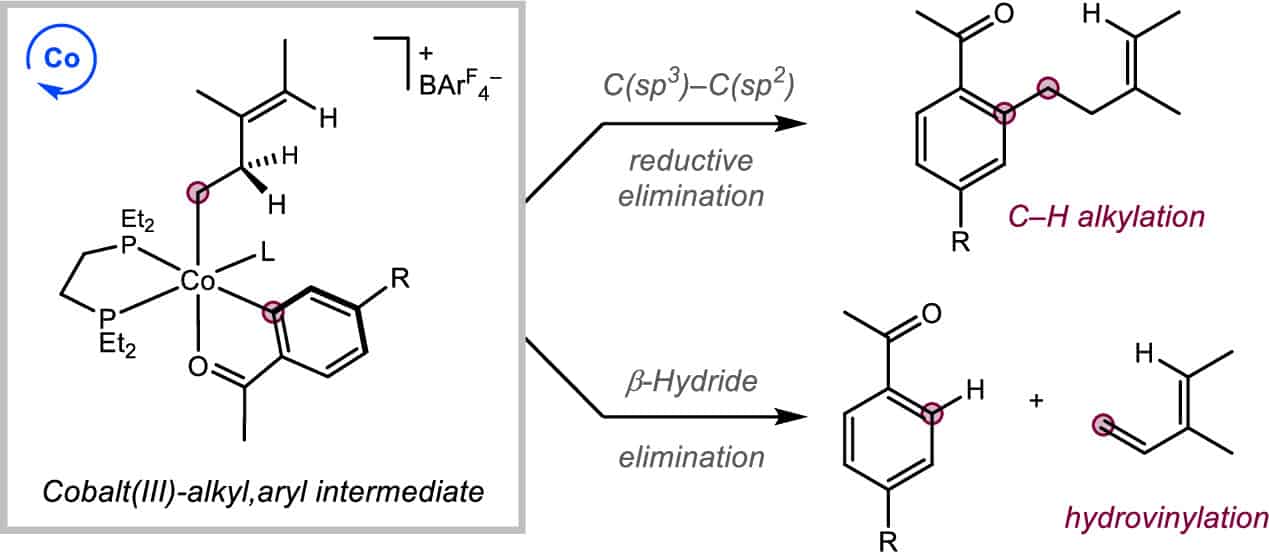 |
246 | Asymmetric Hydrogenation of Indazole-Containing Enamides Relevant to the Synthesis of Zavegepant Using Neutral and Cationic Cobalt PrecatalystsLauren N. Mendelsohn, Connor S. MacNeil, Madison R. Esposito, Tyler P. Pabst, David K. Leahy, Ian W. Davies, and Paul J. Chirik Org. Lett. 2024, 26, 14, 2718–2723 AbstractThe cobalt-catalyzed asymmetric hydrogenation of indazole-containing enamides relevant to the synthesis of the calcitonin gene-related peptide (CGRP) receptor antagonist, zavegepant (1), approved for the treatment of migraines, is described. Both neutral bis(phosphine)cobalt(II) and cationic bis(phosphine)cobalt(I) complexes served as efficient precatalysts for the enamide hydrogenation reactions, providing excellent yield and enantioselectivities (up to >99.9%) for a range of related substrates, though key reactivity differences were observed. Hydrogenation of indazole-containing enamide, methyl (Z)-2-acetamido-3-(7-methyl-1H-indazol-5-yl)acrylate, was performed on a 20 g scale. | 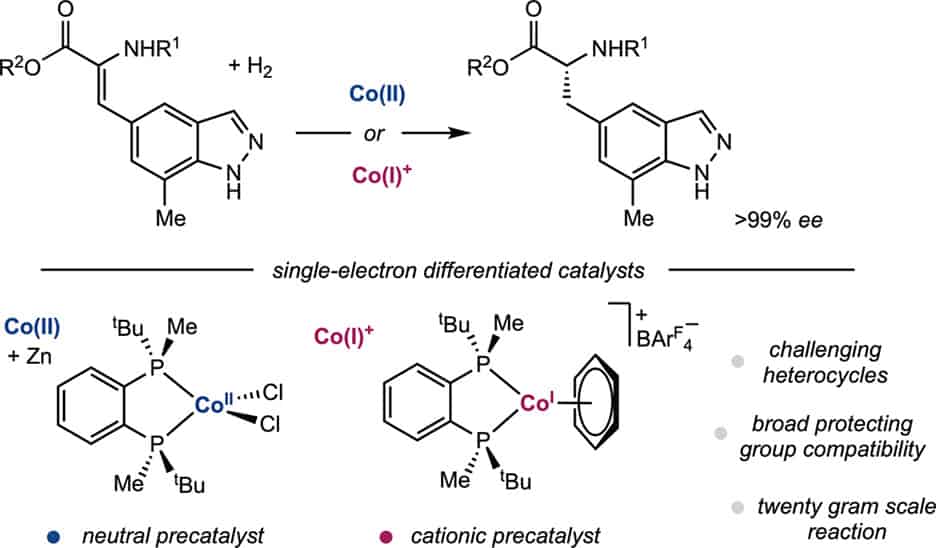 |
245 | Exploring the Effect of Pincer Rigidity on Oxidative Addition Reactions with Cobalt(I) ComplexesBoran Lee, Tyler P. Pabst, Gabriele Hierlmeier, and Paul J. Chirik Organometallics 2023, 42, 8, 708–718 AbstractCobalt complexes containing the 2,6-diaminopyridine-substituted PNP pincer (iPrPNMeNP = 2,6-(iPr2PNMe)2(C5H3N)) were synthesized. A combination of solid-state structures and investigation of the cobalt(I)/(II) redox potentials established a relatively rigid and electron-donating chelating ligand as compared to iPrPNP (iPrPNP = 2,6-(iPr2PCH2)2(C5H3N)). Based on a buried volume analysis, the two pincer ligands are sterically indistinguishable. Nearly planar, diamagnetic, four-coordinate complexes were observed independent of the field strength (chloride, alkyl, aryl) of the fourth ligand completing the coordination sphere of the metal. Computational studies supported a higher barrier for C–H oxidative addition, largely a result of the increased rigidity of the pincer. The increased oxidative addition barrier resulted in stabilization of (iPrPNMeNP)Co(I) complexes, enabling the characterization of the cobalt boryl and the cobalt hydride dimer by X-ray crystallography. Moreover, (iPrPNMeNP)CoMe served as an efficient precatalyst for alkene hydroboration likely because of the reduced propensity to undergo oxidative addition, demonstrating that reactivity and catalytic performance can be tuned by rigidity of pincer ligands. | 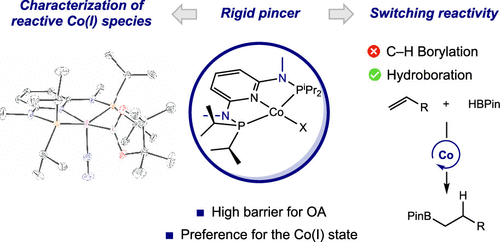 |
244 | Pentamethylcyclopentadienyl Metalloradical Iron Complexes Containing Redox Noninnocent α-Diimine-Type Ligands: Synthesis, Molecular, and Electronic StructuresYoonsu Park, Hongyu Zhong, Tyler P. Pabst, Junho Kim, and Paul J. Chirik Organometallics 2023, 42, 6, 465–472 AbstractThe synthesis and characterization of pentamethylcyclopentadienyl iron complexes bearing the redox noninnocent α-diimine (N,N′-dimesitylbutane-2,3-diimine, MesDI) and α-iminopyridine (N-mesityl(pyridin-2-yl)ethanimine MesPI) ligands were explored. One-pot reduction and complexation of the cyclopentadienyl ring was accomplished by treatment of (κ2–N,N′)FeCl2 (N,N′= MesDI or MesPI) precursors with [C5Me5]Li. The resulting iron compounds were characterized by paramagnetic 1H NMR spectroscopy, magnetic susceptibility measurements, zero-field 57Fe Mössbauer spectroscopy, low-temperature electron paramagnetic resonance (EPR) spectroscopy, and cyclic voltammetry. The combined spectroscopic, structural, and density functional theory (DFT) computational data supported low-spin iron(III) compounds (SFe = 1/2) with π-acidic, formally doubly reduced chelating ligands. |  |
243 | Collaboration as a Key to Advance Capabilities for Earth-Abundant Metal CatalysisPaul J. Chirik, Keary M. Engle, Eric M. Simmons, and Steven R. Wisniewski Org. Process Res. Dev. 2023, 27, 7, 1160–1184 AbstractEarth-abundant metal (EAM) catalysis can have profound impact in the pharmaceutical industry in terms of sustainability and cost improvements from replacing precious metals like palladium as well as harnessing the differential reactivity of first-row metals that allows for novel transformations to enable more efficient routes to clinical candidates. The strategy for building these capabilities within the process group at Bristol Myers Squibb is described herein, with the general plan of building a reaction screening platform, demonstrating scalability, and increasing mechanistic understanding of the reaction and catalyst activation. The development of catalytic transformations utilizing nickel, cobalt, and iron is described while highlighting the importance of collaboration with internal and external groups to advance EAM catalysis and impact our portfolio. The challenges and benefits of working with first-row transition metals, including metrics for the implementation of EAM catalysis, such as cost, process mass intensity, and commercial availability of catalysts and ligands, are discussed. |  |
242 | Bimolecular Reductive Elimination of Ethane from Pyridine(diimine) Iron Methyl Complexes: Mechanism, Electronic Structure, and Entry into [2+2] Cycloaddition CatalysisCarli B. Kovel, Jonathan M. Darmon, S. Chantal E. Stieber, Gisselle Pombar, Tyler P. Pabst, Bastian Theis, Zoë R. Turner, Ökten Üngör, Michael Shatruk, Serena DeBeer, and Paul J. Chirik J. Am. Chem. Soc. 2023, 145, 9, 5061–5073 AbstractThe application of bimolecular reductive elimination to the activation of iron catalysts for alkene–diene cycloaddition is described. Key to this approach was the synthesis, characterization, electronic structure determination, and ultimately solution stability of a family of pyridine(diimine) iron methyl complexes with diverse steric properties and electronic ground states. Both the aryl-substituted, (MePDI)FeCH3 and (EtPDI)FeCH3 (RPDI = 2,6-(2,6-R2-C6H3N═CMe)2C5H3N), and the alkyl-substituted examples, (CyAPDI)FeCH3 (CyAPDI = 2,6-(C6H11N═CMe)2C5H3N), have molecular structures significantly distorted from planarity and S = 3/2 ground states. The related N-arylated derivative bearing 2,6-di-isopropyl aryl substituents, (iPrPDI)FeCH3, has an idealized planar geometry and exhibits spin crossover behavior from S = 1/2 to S = 3/2 states. At 23 °C under an N2 atmosphere, both (MePDI)FeCH3 and (EtPDI)FeCH3 underwent reductive elimination of ethane to form the iron dinitrogen precatalysts, [(MePDI)Fe(N2)]2(μ-N2) and [(EtPDI)Fe(N2)]2(μ-N2), respectively, while (iPrPDI)FeCH3 proved inert to C–C bond formation. By contrast, addition of butadiene to all three iron methyl complexes induced ethane formation and generated the corresponding iron butadiene complexes, (RPDI)Fe(η4-C4H6) (R = Me, Et, iPr), known precatalysts for the [2+2] cycloaddition of olefins and dienes. Kinetic, crossover experiments, and structural studies were combined with magnetic measurements and Mössbauer spectroscopy to elucidate the electronic and steric features of the iron complexes that enable this unusual reductive elimination and precatalyst activation pathway. Transmetalation of methyl groups between iron centers was fast at ambient temperature and independent of steric environment or spin state, while the intermediate dimer underwent the sterically controlled rate-determining reaction with either N2 or butadiene to access a catalytically active iron compound. | ![Bimolecular Reductive Elimination of Ethane from Pyridine(diimine) Iron Methyl Complexes: Mechanism, Electronic Structure, and Entry into [2+2] Cycloaddition Catalysis](/wp-content/uploads/2023/03/img-publ-242.webp) |
241 | Iron-Catalyzed C(sp2)–C(sp3) Suzuki–Miyaura Cross-Coupling Using an Alkoxide BasePaul O. Peterson, Matthew V. Joannou, Eric M. Simmons, Steven R. Wisniewski, Junho Kim, and Paul J. Chirik ACS Catal. 2023, 13, 4, 2443–2448 AbstractThe synthesis and characterization of phenoxy(imine) iron(II) alkyl precatalysts for C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling of aryl boronic esters and alkyl bromides is described. Addition of phenoxyimines (FI) to (py)2Fe(CH2SiMe3)2 (py = pyridine) afforded the high-spin iron(II) alkyl derivatives, (FI)Fe(CH2SiMe3)(py) with varying N-imine substituents. With both neopentyl glycol-protected (BNeo) and pinacol-protected boronic ester (BPin) aryl nucleophiles, an iron-catalyzed cross-coupling method was realized that utilizes mild alkoxide bases. Optimal performance was observed in nonpolar solvents with anisole and fluorobenzene identified as more benign alternatives to benzene. The scope of this transformation includes high efficiency C(sp2)–C(sp3) bond formation with both primary and secondary alkyl bromides with electron-deficient aryl and heteroaryl nucleophiles. Substrates with base-sensitive functionality including ester and nitrile groups were tolerated, highlighting the broader compatibility with an alkoxide base. Radical clock experiments support the formation of electrophile-derived radicals during catalysis, and experiments with preformed potassium aryl boronates demonstrate the role of boronate intermediates in transmetalation. | 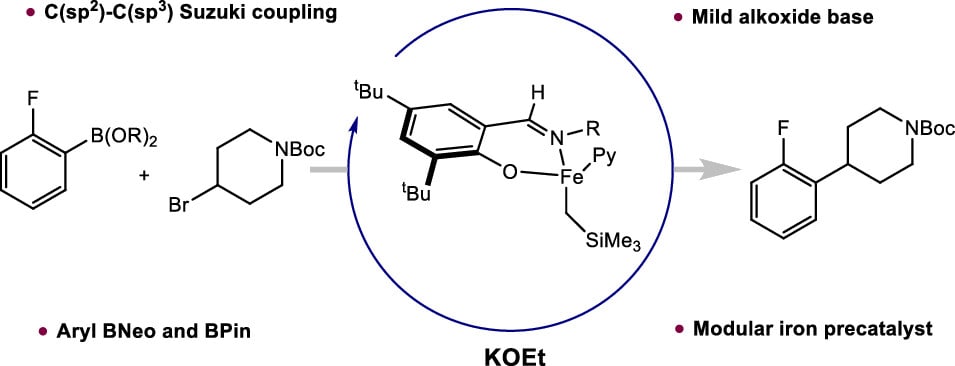 |
240 | Identification of Cyclohexadienyl Hydrides as Intermediates in Molybdenum-Catalyzed Arene HydrogenationGabriele Hierlmeier, Paolo Tosatti, Kurt Puentener, Paul James Chirik Angew. Chem. Int. Ed. 2023, 62, e202216026 AbstractTreatment of phosphino(imino)pyridine (PIP) molybdenum cyclooctadiene (COD) complexes [(PIP)Mo(COD)] with dihydrogen in the presence of benzene selectively furnished the molybdenum cyclohexadienyl hydrides [(PIP)MoH(η5-C6H7)], which are precatalysts for the hydrogenation of benzene to cyclohexane. [(PIP)MoH(η5-C6H7)] arises from a rarely observed insertion of benzene into a molybdenum–hydride bond, a key step in the molybdenum-catalyzed homogeneous hydrogenation of arenes. The reaction with toluene afforded a single isomer of the corresponding molybdenum cyclohexadienyl hydride while para-xylene instead predominantly formed the molybdenum η6-arene complex with the insertion product being a minor component. Addition of carbon monoxide to a cyclohexane-d12 solution of [(PIP)MoH(η5-C6H7)] liberated cyclohexadiene, providing experimental support for a higher kinetic barrier for the subsequent steps en route to cycloalkanes. |  |
239 | Catalytic N–H Bond Formation Promoted by a Ruthenium Hydride Complex Bearing a Redox-Active Pyrimidine-Imine LigandSangmin Kim, Junho Kim, Hongyu Zhong, Grace B. Panetti, and Paul J. Chirik J. Am. Chem. Soc. 2022, 144, 45, 20661–20671 AbstractThe synthesis of a piano-stool ruthenium hydride, [(η5-C5Me5)Ru(PmIm)H] (PmIm = (N-(1,3,5-trimethylphenyl)-1-(pyrimidin-2-yl)ethan-1-imine), for the dual purpose of catalytic dihydrogen activation and subsequent hydrogen atom transfer for the formation of weak chemical bonds is described. The introduction of a neutral, potentially redox-active PmIm supporting ligand was designed to eliminate the possibility of deleterious C(sp2)–H reductive coupling and elimination that has been identified as a deactivation pathway with related rhodium and iridium catalysts. Treatment of [(η5-C5Me5)RuCl2]n with one equivalent PmIm ligand in the presence of zinc and sodium methoxide resulted in the isolation of the diruthenium complex, [(η5-C5Me5)Ru(PmIm)]2, arising from the C–C bond formation between two PmIm chelates. Addition of H2 to the ruthenium dimer under both thermal and blue light irradiation conditions furnished the targeted hydride, [(η5-C5Me5)Ru(PmIm)H], which has a relatively weak DFT-calculated Ru–H bond dissociation free energy (BDFE) of 47.9 kcal/mol. Addition of TEMPO to [(η5-C5Me5)Ru(PmIm)H] generated the 17-electron metalloradical, [(η5-C5Me5)Ru(PmIm)], which was characterized by EPR spectroscopy. The C–C bond forming process was reversible as the irradiation of [(η5-C5Me5)Ru(PmIm)]2 generated [(η5-C5Me5)Ru(PmIm)H] and a piano-stool ruthenium complex containing an enamide ligand derived from H-atom abstraction from the PmIm chelate. Equilibration studies were used to establish an experimental estimate of the effective Ru–H BDFE, and a value of 50.8 kcal/mol was obtained, in agreement with the observed loss of H2 and the DFT-computed value. The ruthenium hydride was an effective catalyst for the thermal catalytic hydrogenation of TEMPO, acridine, and a cobalt-imido complex and for the selective reduction of azobenzene to diphenylhydrazine, highlighting the role of this complex in catalytic weak bond formation using H2 as the stoichiometric reductant. | 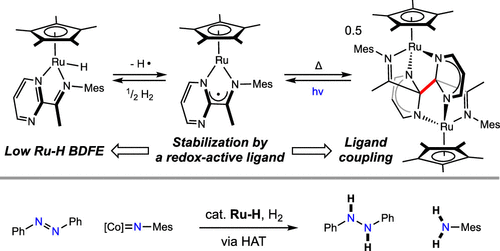 |
238 | Alcohol Synthesis by Cobalt-Catalyzed Visible-Light-Driven Reductive HydroformylationConnor S. MacNeil, Lauren N. Mendelsohn, Tyler P. Pabst, Gabriele Hierlmeier, and Paul J. Chirik J. Am. Chem. Soc. 2022, 144, 42, 19219–19224 AbstractA cobalt-catalyzed reductive hydroformylation of terminal and 1,1-disubstituted alkenes is described. One-carbon homologated alcohols were synthesized directly from CO and H2, affording anti-Markovnikov products (34–87% yield) with exclusive regiocontrol (linear/branch >99:1) for minimally functionalized alkenes. Irradiation of the air-stable cobalt hydride, (dcype)Co(CO)2H (dcype = dicyclohexylphosphinoethane) with blue light generated the active catalyst that mediates alkene hydroformylation and subsequent aldehyde hydrogenation. Mechanistic origins of absolute regiocontrol were investigated by in situ monitoring of the tandem catalytic reaction using multinuclear NMR spectroscopy with syngas mixtures. |  |
237 | C–H Activation by Isolable Cationic Bis(phosphine) Cobalt(III) MetallacyclesWilliam G. Whitehurst, Junho Kim, Stefan G. Koenig, and Paul J. Chirik J. Am. Chem. Soc. 2022, 144, 41, 19186–19195 AbstractFive- and six-coordinate cationic bis(phosphine) cobalt(III) metallacycle complexes were synthesized with the general structures, [(depe)Co(cycloneophyl)(L)(L′)][BArF4] (depe = 1,2-bis(diethylphosphino)ethane; cycloneophyl = [κ-C:C-(CH2C(Me)2)C6H4]; L/L′ = pyridine, pivalonitrile, or the vacant site, BAr4F = B[(3,5-(CF3)2)C6H3]4). Each of these compounds promoted facile directed C(sp2)–H activation with exclusive selectivity for ortho-alkylated products, consistent with the selectivity of reported cobalt-catalyzed arene-alkene-alkyne coupling reactions. The direct observation of C–H activation by cobalt(III) metallacycles provided experimental support for the intermediacy of these compounds in this class of catalytic C–H functionalization reaction. Deuterium labeling and kinetic studies provided insight into the nature of C–H bond cleavage and C–C bond reductive elimination from isolable cobalt(III) precursors. |  |
236 | Mechanistic Investigations of the Asymmetric Hydrogenation of Enamides with Neutral Bis(phosphine) Cobalt PrecatalystsLauren N. Mendelsohn, Ljiljana Pavlovic, Hongyu Zhong, Max R. Friedend, Michael Shevlin, Kathrin H. Hopmann, and Paul J. Chirik J. Am. Chem. Soc. 2022, 144, 34, 15764–15778 AbstractThe mechanism of the asymmetric hydrogenation of prochiral enamides by well-defined, neutral bis(phosphine) cobalt(0) and cobalt(II) precatalysts has been explored using(R,R)-iPrDuPhos ((R,R)-iPrDuPhos = (+)-1,2-bis[(2R,5R)-2,5-diisopropylphospholano]benzene) as a representative chiral bis(phosphine) ligand. A series of (R,R)-(iPrDuPhos)Co(enamide) (enamide = methyl-2-acetamidoacrylate (MAA), methyl(Z)-α-acetamidocinnamate (MAC), and methyl(Z)-acetamido(4-fluorophenyl)acrylate (4FMAC)) complexes (1-MAA, 1-MAC, and 1-4FMAC), as well as a dinuclear cobalt tetrahydride, [(R,R)-(iPrDuPhos)Co]2(μ2-H)3(H) (2), were independently synthesized, characterized, and evaluated in both stoichiometric and catalytic hydrogenation reactions. Characterization of (R,R)-(iPrDuPhos)Co(enamide) complexes by X-ray diffraction established the formation of the pro-(R) diastereomers in contrast to the (S)-alkane products obtained from the catalytic reaction. In situ monitoring of the cobalt-catalyzed hydrogenation reactions by UV–visible and freeze-quench electron paramagnetic resonance spectroscopies revealed (R,R)-(iPrDuPhos)Co(enamide) complexes as the catalyst resting state for all the three enamides studied. Variable time normalization analysis kinetic studies of the cobalt-catalyzed hydrogenation reactions in methanol established a rate law that is first order in (R,R)-(iPrDuPhos)Co(enamide) and H2 but independent of the enamide concentration. Deuterium-labeling studies, including measurement of an H2/D2 kinetic isotope effect and catalytic hydrogenations with HD, established an irreversible H2 addition step to the bound enamide. Density functional theory calculations support that this step is both rate and selectivity determining. Calculations, as well as HD-labeling studies, provide evidence for two-electron redox cycling involving cobalt(0) and cobalt(II) intermediates during the catalytic cycle. Taken together, these experiments support an unsaturated pathway for the [(R,R)-(iPrDuPhos)Co]-catalyzed hydrogenation of prochiral enamides. |  |
235 | Cobalt-Catalyzed Asymmetric Hydrogenation of Enamides: Insights into Mechanisms and Solvent EffectsLjiljana Pavlovic, Lauren N. Mendelsohn, Hongyu Zhong, Paul J. Chirik, and Kathrin H. Hopmann Organometallics 2022, 41, 1872–1882. AbstractThe mechanistic details of the (PhBPE)Co-catalyzed asymmetric hydrogenation of enamides are investigated using computational and experimental approaches. Four mechanistic possibilities are compared: a direct Co(0)/Co(II) redox path, a metathesis pathway, a nonredox Co(II) mechanism featuring an aza-metallacycle, and a possible enamide–imine tautomerization pathway. The results indicate that the operative mechanism may depend on the type of enamide. Explicit solvent is found to be crucial for the stabilization of transition states and for a proper estimation of the enantiomeric excess. The combined results highlight the complexity of base-metal-catalyzed hydrogenations but do also provide guiding principles for a mechanistic understanding of these systems, where protic substrates can be expected to open up nonredox hydrogenation pathways. |  |
234 | Nickel-Catalyzed Dimerization of Di- and Trisubstituted OlefinsSam Yruegas, Michele Paccagnini, Suzzy C. Ho, Aaron Sattler, and Paul J. Chirik Organometallics 2022, 41, 2059–2066. AbstractThe dimerization of a series of di- and trisubstituted C5-olefins catalyzed by hexafluoroacetylacetonate (acacCF3) nickel complexes is described. The dimerization of the trisubstituted olefin 2-methyl-2-butene by both a single-component nickel precatalyst and in situ activation of a bis(acacCF3) nickel precatalyst produced predominantly C10-dimers with nearly identical product distributions. The dimers produced by the nickel catalysts are less-branched than acid-catalyzed (e.g., BF3) dimers, due to the ability of the nickel intermediate to function as both an olefin isomerization and a dimerization catalyst, whereby C–C bond formation occurs preferentially between a primary carbon of one olefin and the less substituted carbon of the inserting olefin. The dimerization of the regioisomers of 2-methyl-2-butene, 2-methyl-1-butene, and 3-methyl-1-butene, utilizing the aforementioned nickel precatalysts, resulted in the same structural isomers of the dimers but different product distributions, indicating that dimerization occurred at similar rate or faster than isomerization. Stoichiometric experiments, product analysis, and observations from in situ nuclear magnetic resonance spectroscopy support a mechanism whereby a short-lived nickel-hydride intermediate promotes chain insertion, chain walking, and isomerization to give less-branched dimers from highly substituted olefins than would be achieved with acid-catalyzed dimerization. |  |
233 | C(sp2)–H Activation with Bis(silylene)pyridine Cobalt(III) Complexes: Catalytic Hydrogen Isotope Exchange of Sterically Hindered C–H BondsJose B. Roque, Tyler P. Pabst, and Paul J. Chirik ACS Catal. 2022, 12, 8877–8885. AbstractThe bis(silylene)pyridine cobalt(III) dihydride boryl, trans-[ptolSiNSi]Co(H)2BPin (ptolSiNSi = 2,6-[EtNSi(NtBu)2CAr]2 C5H3N, where ptol = 4-MeC6H4, and Pin = pinacolato) has been used as a precatalyst for the hydrogen isotope exchange (HIE) of arenes and heteroarenes using benzene-d6 as the deuterium source. Use of D2 as the source of the isotope produced modest levels of deuterium incorporation, and stoichiometric studies established modification of the pincer ligand through irreversible addition of H2 across the silylene leading to catalyst deactivation. High levels of deuterium incorporation were observed with benzene-d6 as the isotope source and enabled low (0.5–5 mol %) loadings of the cobalt precursor. The resulting high activity for C–H activation resulted in deuterium incorporation at sterically encumbered sites previously inaccessible with first-row metal HIE catalysts. The cobalt-catalyzed method was also compatible with aryl halides, demonstrating a kinetic preference for chemoselective C(sp2)–H activation over C(sp2)–X (X = Cl, Br) bonds. Monitoring the catalytic reaction by NMR spectroscopy established cobalt(III) resting states at both low and high conversions of the substrate, and the overall performance was inhibited by the addition of HBPin. Studies on precatalyst activation with cis-[ptolSiNSi]Co(Bf)2H and cis-[ptolSiNSi]Co(H)2Bf (where Bf = 2-benzofuranyl) support the intermediacy of bis(hydride)aryl cobalt intermediates as opposed to bis(aryl)hydride cobalt complexes in the catalytic HIE method. Mechanistic insights resulted in an improved protocol using [ptolSiNSi]Co(H)3·NaBHEt3 as the precatalyst, ultimately translating onto higher levels of isotopic incorporation. |  |
232 | Molybdenum-Catalyzed Asymmetric Hydrogenation of Fused Arenes and HeteroarenesPeter Viereck, Gabriele Hierlmeier, Paolo Tosatti, Tyler P. Pabst, Kurt Puentener, and Paul J. Chirik J. Am. Chem. Soc. 2022, 144, 11203–11214. AbstractThe synthesis of enantioenriched molybdenum precatalysts for the asymmetric hydrogenation of substituted quinolines and naphthalenes is described. Three classes of pincer ligands with chiral substituents were evaluated as supporting ligands in the molybdenum-catalyzed hydrogenation reactions, where oxazoline imino(pyridine) chelates were identified as optimal. A series of 2,6-disubstituted quinolines was hydrogenated to enantioenriched decahydroquinolines with high diastereo- and enantioselectivities. For quinoline derivatives, selective hydrogenation of both the carbocycle and heterocycle was observed depending on the ring substitution. Spectroscopic and mechanistic studies established molybdenum η6-arene complexes as the catalyst resting state and that partial hydrogenation arises from dissociation of the substrate from the coordination sphere of molybdenum prior to complete reduction. A stereochemical model is proposed based on the relative energies of the respective coordination of the prochiral faces of the arene determined by steric interactions between the substrate and the chiral ligand, rather than through precoordination by a heteroatom. |  |
231 | Cationic Bis(phosphine) Cobalt(I) Arene Complexes as Precatalysts for the Asymmetric Synthesis of SitagliptinConnor S. MacNeil, Hongyu Zhong, Tyler P. Pabst, Michael Shevlin, and Paul J. Chirik ACS Catal. 2022, 12, 4680–4687. AbstractThe cobalt-catalyzed asymmetric hydrogenation of dehydro-sitagliptin was studied and applied to the synthesis of sitagliptin (Januvia). Catalyst discovery efforts were accelerated by the application of a general method for the synthesis of cationic bis(phosphine) cobalt η6-arene complexes. One-electron oxidation of bis(phosphine) cobalt(II) dialkyl complexes in the presence of arenes furnished the corresponding, bench-stable cobalt precatalysts, [(P–P)Co(η6-C6H6)][BAr4F]. Asymmetric hydrogenation utilized 0.5 mol % of the optimal catalyst, [(R,R)-(iPrDuPhos)Co(η6-C6H6)][BAr4F], in THF solution and produced sitagliptin in >99% yield with 97% ee. Cobalt catalysts were compatible with a range of solvents and maintained excellent activity and selectivity after standing in air in the solid state for 2 weeks. Deuterium labeling studies support an enamine–imine tautomerization process resulting in the reduction of the metalated imine. Notably, state-of-the-art neutral bis(phosphine) cobalt precatalysts were ineffective, emphasizing the utility of a class of cationic cobalt precatalysts. |  |
230 | Development of Cobalt Catalysts for the meta-Selective C(sp2)–H Borylation of Fluorinated ArenesTyler P. Pabst and Paul J. Chirik J. Am. Chem. Soc. 2022, 144, 6465–6474. AbstractCobalt precatalysts for the meta-selective borylation of fluorinated arenes are described. Initial screening and stoichiometric reactivity studies culminated in the preparation of a cobalt alkyl precatalyst supported by the sterically protected terpyridine (5,5″-Me2ArTpy = 4′-(4-N,N′-dimethylaminophenyl)-5,5″-dimethyl-2,2′:6′,2″-terpyridine). Under the optimized conditions, borylation with this precatalyst afforded up to 16 turnovers and near-exclusive meta regioselectivity with a range of substituted fluoroarenes in cyclopentyl methyl ether solvent at room temperature. Deuterium kinetic isotope effects of 2.9(2) at 23 °C support a turnover-limiting and selectivity-determining C(sp2)–H activation step, and stoichiometric C–H activation experiments provided insights into the identity of the C–H activating intermediate in catalysis. Analysis of the relevant Co–C and C–H bond thermodynamics support that the thermodynamics of C–H activation favor ortho-to-fluorine selectivity, providing additional, indirect support for kinetic control of C–H activation as the origin of meta selectivity. | 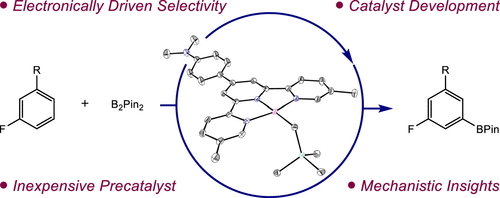 |
229 | Ammonia synthesis by photocatalytic hydrogenation of a N2-derived molybdenum nitrideSangmin Kim, Yoonsu Park, Junho Kim, Tyler P. Pabst, and Paul J. Chirik Nature Synthesis 2022, 1, 297–303 AbstractAlthough metal complexes are known to split dinitrogen at ambient temperature and pressure, the synthesis of ammonia from these compounds with H2 as the terminal reductant is rarely achieved. Here we report a photocatalytic ammonia synthesis from a N2-derived terminal molybdenum nitride by using H2 as the terminal reductant. An iridium hydride photocatalyst mediates the reaction on irradiation with blue light. A molybdenum pentahydride was identified as the principal metal product to arise after ammonia release. Conversion of the molybdenum pentahydride back to the terminal molybdenum nitride was accomplished in three steps and completed a synthetic cycle for NH3 formation from N2 and H2. Mechanistic investigations support a pathway that involves photoexcitation of the iridium hydride and a subsequent energy transfer rather than electron transfer. Deuterium labelling confirmed H2 as the source of the N–H bonds. This photodriven, proton-coupled electron transfer allows the use of H2 as the terminal reductant for the catalytic formation of NH3 from N2 using metal catalysts. |  |
228 | Three-Component Coupling of Arenes, Ethylene, and Alkynes Catalyzed by a Cationic Bis(phosphine) Cobalt Complex: Intercepting Metallacyclopentenes for C–H FunctionalizationWilliam G. Whitehurst, Junho Kim, Stefan G. Koenig, and Paul J. Chirik J. Am. Chem. Soc. 2022, 144, 4530–4540. AbstractA cobalt-catalyzed intermolecular three-component coupling of arenes, ethylene, and alkynes was developed using the well-defined air-stable cationic bis(phosphine) cobalt(I) complex, [(dcype)Co(η6-C7H8)][BArF4] (dcype = 1,2-bis(dicyclohexylphosphino)ethane; BArF4 = B[(3,5-(CF3)2)C6H3]4), as the precatalyst. All three components were required for turnover and formation of ortho-homoallylated arene products. A range of directing groups including amide, ketone, and 2-pyridyl substituents on the arene promoted the reaction. The cobalt-catalyzed method exhibited broad functional group tolerance allowing for the late-stage functionalization of two drug molecules, fenofibrate and haloperidol. A series of control reactions, deuterium labeling studies, resting state analysis, as well as synthesis of substrate- and product-bound η6-arene complexes supported a pathway involving C(sp2)–H activation from a cobalt(III) metallacycle. | 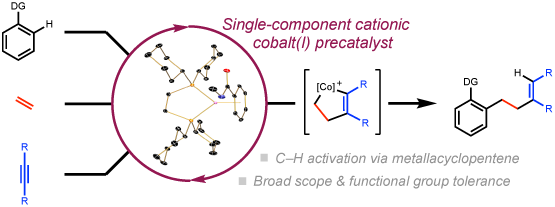 |
227 | (PNP)Cobalt-Catalyzed Olefination of DiazoalkanesSam Yruegas, Scott P. Semproni, and Paul J. Chirik AbstractAddition of excess diazoalkane to the pincer-supported cobalt(I) dinitrogen complex (tBumPNP)CoN2 (tBumPNP = modified 2,6-bis[(ditert-butylphosphino)methyl]pyridine) resulted in the catalytic formation of the homocoupled alkene product with concomitant loss of N2. Monosubstituted diazoalkanes, trimethylsilyldiazomethane and tolyldiazomethane, generated the olefin product in quantitative yield with exclusive (E)-stereoselectivity. Disubstituted diazoalkanes, diphenyldiazomethane and 9-diazofluorene, also yielded the olefin as the major product along with minor azine coupling. Investigations into the nature of the diazoalkane–cobalt interaction by multinuclear NMR spectroscopy and X-ray diffraction established end-on diazoalkane cobalt complexes as the resting states. The isolated four-coordinate cobalt diazoalkane complexes promoted conversion to the corresponding olefin. The reaction of (tBumPNP)CoN2 with an α-diazo-β-ketoester resulted in the formation of a five-coordinate Co(I)-diazoalkane complex with a chelating ester unit that was unreactive for olefination. | 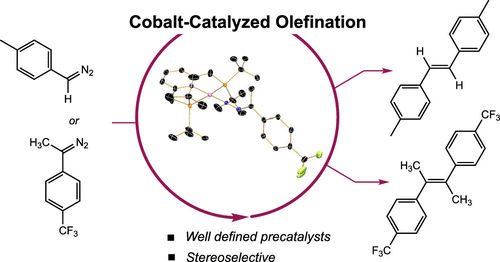 |
226 | Visible-Light-Driven, Iridium-Catalyzed Hydrogen Transfer: Mechanistic Studies, Identification of Intermediates, and Catalyst ImprovementsYoonsu Park, Lei Tian, Sangmin Kim, Tyler P. Pabst, Junho Kim, Gregory D. Scholes, and Paul J. Chirik AbstractThe harvesting of visible light is a powerful strategy for the synthesis of weak chemical bonds involving hydrogen that are below the thermodynamic threshold for spontaneous H2 evolution. Piano-stool iridium hydride complexes are effective for the blue-light-driven hydrogenation of organic substrates and contra-thermodynamic dearomative isomerization. In this work, a combination of spectroscopic measurements, isotopic labeling, structure–reactivity relationships, and computational studies has been used to explore the mechanism of these stoichiometric and catalytic reactions. Photophysical measurements on the iridium hydride catalysts demonstrated the generation of long-lived excited states with principally metal-to-ligand charge transfer (MLCT) character. Transient absorption spectroscopic studies with a representative substrate, anthracene revealed a diffusion-controlled dynamic quenching of the MLCT state. The triplet state of anthracene was detected immediately after the quenching events, suggesting that triplet–triplet energy transfer initiated the photocatalytic process. The key role of triplet anthracene on the post-energy transfer step was further demonstrated by employing photocatalytic hydrogenation with a triplet photosensitizer and a HAT agent, hydroquinone. DFT calculations support a concerted hydrogen atom transfer mechanism in lieu of stepwise electron/proton or proton/electron transfer pathways. Kinetic monitoring of the deactivation channel established an inverse kinetic isotope effect, supporting reversible C(sp2)–H reductive coupling followed by rate-limiting ligand dissociation. Mechanistic insights enabled design of a piano-stool iridium hydride catalyst with a rationally modified supporting ligand that exhibited improved photostability under blue light irradiation. The complex also provided improved catalytic performance toward photoinduced hydrogenation with H2 and contra-thermodynamic isomerization. | 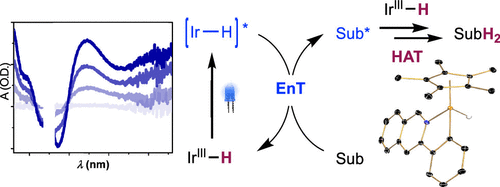 |
225 | Cobalt-Catalyzed C(sp2)–C(sp3) Suzuki-Miyaura Cross-Coupling Enabled by Well-Defined Precatalysts with L,X-Type LigandsL. Reginald Mills, David Gygi, Jacob R. Ludwig, Eric M. Simmons, Steven R. Wisniewski, Junho Kim, and Paul J. Chirik ACS Catal. 2022, 12, 1905–1918. AbstractCobalt(II) halides in combination with phenoxyimine (FI) ligands generated efficient precatalysts in situ for the C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling between alkyl bromides and neopentylglycol (hetero)arylboronic esters. The protocol enabled efficient C–C bond formation with a host of nucleophiles and electrophiles (36 examples, 34–95%) with precatalyst loadings of 5 mol %. Studies with alkyl halide electrophiles that function as radical clocks support the intermediacy of alkyl radicals during the course of the catalytic reaction. The improved performance of the FI–cobalt catalyst was correlated with decreased lifetimes of cage-escaped radicals as compared to those of diamine-type ligands. Studies of the phenoxyimine–cobalt coordination chemistry validate the L,X interaction leading to the discovery of an optimal, well-defined, air-stable mono-FI–cobalt(II) precatalyst structure. |  |
224 | Effect of Pincer Methylation on the Selectivity and Activity in (PNP)Cobalt-Catalyzed C(sp2)–H BorylationBoran Lee, Tyler P. Pabst, and Paul J. Chirik Organometallics 2021, 40, 3766–3774. AbstractCobalt complexes supported by a tetramethylated PNP pincer ligand (Me4iPrPNP = 2,6-(iPr2PCMe2)2(C5H3N)) have been synthesized and structurally characterized. Examples include cobalt(I)–choride, −methyl, −aryl, and −benzofuranyl derivatives. The performance of these compounds was evaluated in the catalytic borylation of fluorinated arenes using B2Pin2 as the boron source. While P–C bond cleavage, a known deactivation pathway in [(PNP)Co]-catalyzed borylation, was suppressed, the overall activity and selectivity of the borylation of fluoroarenes was reduced as compared to the previously reported [(PNP)Co] catalyst lacking isopropylene spacers. Stoichiometric reactions support an increased barrier for oxidative addition to cobalt(I), a result of the increased steric profile and decreased conformational flexibility of the pincer resulting from methylation distal to the active site. With a more activated substrate such as benzofuran, catalytic borylation with cobalt(I) precatalysts and HBPin was observed. Monitoring the progress of the reaction by NMR spectroscopy revealed the presence of cobalt(III) intermediates during the course of the borylation, supporting a cobalt(I)-(III) redox cycle. |  |
223 | Well-Defined Cationic Cobalt(I) Precatalyst for Olefin-Alkyne [2+2] Cycloaddition and Olefin-Diene Hydrovinylation Reactions: Experimental Evidence for Metallacycle IntermediatesMarcus E. Farmer, Lauren E. Ehehalt, Tyler P. Pabst, Matthew T. Tudge, and Paul J. Chirik Organometallics 2021, 40, 3599–3607. AbstractThe synthesis and characterization of the cationic cobalt(I) arene complex, [(dppf)Co(η6-C7H8)][BArF4] (dppf = 1,1′-bis(diphenylphosphino)ferrocene; BArF4 = B[(3,5-(CF3)2)C6H3]4) from an air-stable cobalt precursor is described. Dissolution in benzene-d6 or tetrahydrofuran (THF) resulted in rapid arene substitution and generated [(dppf)Co(η6-C6H6)][BArF4] or [(dppf)Co(THF)2][BArF4]. The latter compound was characterized by a combination of X-ray diffraction and magnetometry and established an S = 1 cobalt(I) derivative. The isolated bis(phosphine)cobalt complexes were evaluated as precatalysts for carbon–carbon bond-forming reactions. The [2 + 2] cycloaddition of internal alkynes and olefins was observed with cobalt precatalyst loadings of 0.25 mol % with high chemoselectivity. The catalytic method was compatible with Lewis basic functional groups, an advantage over in situ-generated catalysts that rely on excess trialkyl aluminum activators. The cationic bis(phosphine)cobalt arene complex was also an effective catalyst precursor for the hydrovinylation of isoprene with ethylene. In both C–C bond-forming reactions, the corresponding cobalt(0) complex, [(dppf)Co(COD)] (COD = 1,5-cyclooctadiene), was inactive, providing strong evidence of the role of cobalt(I) during catalysis. In both catalytic reactions, deuterium crossover experiments provide experimental evidence of the role of metallacyclic intermediates during turnover. | ![Well-Defined Cationic Cobalt(I) Precatalyst for Olefin-Alkyne [2+2] Cycloaddition and Olefin-Diene Hydrovinylation Reactions: Experimental Evidence for Metallacycle Intermediates](/wp-content/uploads/2022/10/img-publ-223.gif) |
222 | Catalyst Design Principles Enabling Intermolecular Alkene-Diene [2+2] Cycloaddition and Depolymerization ReactionsMegan Mohadjer Beromi, Jarod M. Younker, Hongyu Zhong, Tyler P. Pabst, and Paul J. Chirik J. Am. Chem. Soc. 2021, 143, 17793–17805. AbstractAryl-substituted pyridine(diimine) iron complexes promote the catalytic [2 + 2] cycloadditions of alkenes and dienes to form vinylcyclobutanes as well as the oligomerization of butadiene to generate divinyl(oligocyclobutane), a microstructure of poly(butadiene) that is chemically recyclable. A systematic study on a series of iron butadiene complexes as well as their ruthenium congeners has provided insights into the essential features of the catalyst that promotes these cycloaddition reactions. Structural and computational studies on iron butadiene complexes identified that the structural rigidity of the tridentate pincer enables rare s-trans diene coordination. This geometry, in turn, promotes dissociation of one of the alkene arms of the diene, opening a coordination site for the incoming substrate to engage in oxidative cyclization. Studies on ruthenium congeners established that this step occurs without redox involvement of the pyridine(diimine) chelate. Cyclobutane formation occurs from a metallacyclic intermediate by reversible C(sp3)–C(sp3) reductive coupling. A series of labeling experiments with pyridine(diimine) iron and ruthenium complexes support the favorability of accessing the +3 oxidation state to trigger C(sp3)–C(sp3) reductive elimination, involving spin crossover from S = 0 to S = 1. The high density of states of iron and the redox-active pyridine(diimine) ligand facilitate this reactivity under thermal conditions. For the ruthenium congener, the pyridine(diimine) remains redox innocent and irradiation with blue light was required to promote the analogous reactivity. These structure–activity relationships highlight important design principles for the development of next generation catalysts for these cycloaddition reactions as well as the promotion of chemical recycling of cycloaddition polymers. | ![Catalyst Design Principles Enabling Intermolecular Alkene-Diene [2+2] Cycloaddition and Depolymerization Reactions](/wp-content/uploads/2022/10/img-publ-222.gif) |
221 | Visible light enables catalytic formation of weak chemical bonds with molecular hydrogenYoonsu Park, Sangmin Kim, Lei Tian, Hongyu Zhong, Gregory D. Scholes, and Paul J. Chirik AbstractThe synthesis of weak chemical bonds at or near thermodynamic potential is a fundamental challenge in chemistry, with applications ranging from catalysis to biology to energy science. Proton-coupled electron transfer using molecular hydrogen is an attractive strategy for synthesizing weak element–hydrogen bonds, but the intrinsic thermodynamics presents a challenge for reactivity. Here we describe the direct photocatalytic synthesis of extremely weak element–hydrogen bonds of metal amido and metal imido complexes, as well as organic compounds with bond dissociation free energies as low as 31 kcal mol−1. Key to this approach is the bifunctional behaviour of the chromophoric iridium hydride photocatalyst. Activation of molecular hydrogen occurs in the ground state and the resulting iridium hydride harvests visible light to enable spontaneous formation of weak chemical bonds near thermodynamic potential with no by-products. Photophysical and mechanistic studies corroborate radical-based reaction pathways and highlight the uniqueness of this photodriven approach in promoting new catalytic chemistry. | 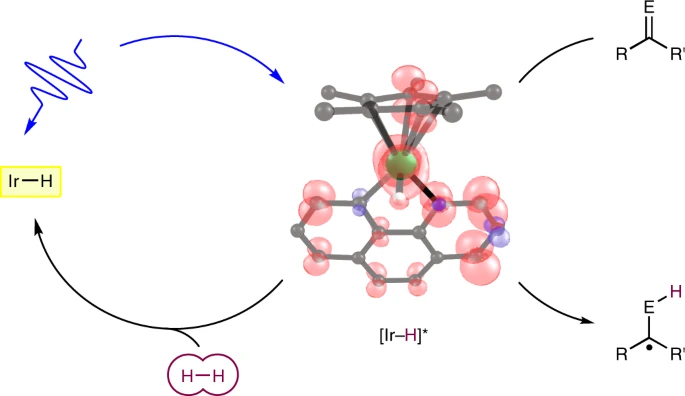 |
220 | Synthesis, Electronic Structure, and Reactivity of a Planar Four-Coordinate, Cobalt–Imido ComplexYoonsu Park, Scott P. Semproni, Hongyu Zhong, and Paul J. Chirik Angew. Chemie Int. Ed. 2021, 60, 14376–14380. AbstractA four-coordinate cobalt–imido complex, (tBumPNP)Co=NMes (tBumPNP=modified PNP pincer ligand) has been synthesized from addition of 2,4,6-trimethylphenylazide (Mes–N3) to the corresponding dinitrogen complex. The solid-state structure determined by X-ray diffraction established a rare, idealized planar geometry with a Co=N bond distance of 1.716(2) Å. Magnetic measurements revealed an S=1 ground state with CAS-SCF calculations supporting radical character on the imide nitrogen. Thermolysis of the cobalt–imido compound induced selective insertion of the imido group into a Co−P bond and yielded a three-coordinate cobalt complex with a distorted T-shaped geometry. Transition state analysis conducted with DFT calculations established the thermodynamic stability of the P–N coupled product and provided insight into the exclusive selectivity. |  |
219 | Oxidative Addition of Aryl and Alkyl Halides to a Reduced Iron Pincer ComplexStephan M. Rummelt, Paul O. Peterson, Hongyu Zhong, and Paul J. Chirik J. Am. Chem. Soc. 2021, 143, 5928–5936. AbstractThe two-electron oxidative addition of aryl and alkyl halides to a reduced iron dinitrogen complex with a strong-field tridentate pincer ligand has been demonstrated. Addition of iodobenzene or bromobenzene to (3,5-Me2MesCNC)Fe(N2)2 (3,5-Me2MesCNC = 2,6-(2,4,6-Me-C6H2-imidazol-2-ylidene)2-3,5-Me2-pyridine) resulted in rapid oxidative addition and formation of the diamagnetic, octahedral Fe(II) products (3,5-Me2MesCNC)Fe(Ph)(N2)(X), where X = I or Br. Competition experiments established the relative rate of oxidative addition of aryl halides as I > Br > Cl. A linear free energy of relative reaction rates of electronically differentiated aryl bromides (ρ = 1.5) was consistent with a concerted-type pathway. The oxidative addition of alkyl halides such as methyl-, isobutyl-, or neopentyl halides was also rapid at room temperature, but substrates with more accessible β-hydrogen positions (e.g., 1-bromobutane) underwent subsequent β-hydride elimination. Cyclization of an alkyl halide containing a radical clock and epimerization of neohexyl iodide-d2 upon oxidative addition to (3,5-Me2MesCNC)Fe(N2)2 are consistent with radical intermediates during C(sp3)–X bond cleavage. Importantly, while C(sp2)–X and C(sp3)–X oxidative addition produces net two-electron chemistry, the preferred pathway for obtaining the products is concerted and stepwise, respectively. | 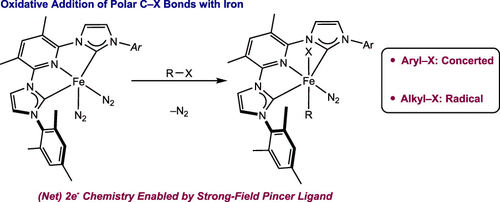 |
218 | Synthesis and Asymmetric Alkene Hydrogenation Activity of C2-Symmetric Enantioenriched Pyridine Dicarbene Iron Dialkyl ComplexesPeter Viereck, Stephan M. Rummelt, Natalia A. Soja, Tyler P. Pabst, and Paul J. Chirik Organometallics 2021, 40, 1053–1061. AbstractEnantioenriched N-alkyl-imidazole-substituted pyridine dicarbene iron dialkyl complexes have been synthesized and characterized by 1H NMR and zero-field 57Fe Mössbauer spectroscopies as well as single-crystal X-ray diffraction. In benzene-d6, reversible coordination of N2 was observed establishing an equilibrium between a five-coordinate, S = 1 iron dialkyl derivative and the corresponding six-coordinate, diamagnetic dinitrogen complex. A modest enantioselectivity of 45% enantiomeric excess (ee) was observed for the catalytic asymmetric hydrogenation of 1-isopropyl-1-phenyl ethylene at 4 atm of H2 using 10 mol % of an enantioenriched iron dialkyl precatalyst, (ACNC)Fe(CH2SiMe3)2 ((ACNC) = bis(alkylimidazol-2-ylidene)pyridine). Decreasing the H2 pressure to 1 atm increased the ee to 70%. Incubation experiments established that the reaction of the iron dialkyl precatalysts with H2 initiates a background reaction leading to the generation of a less selective catalyst; suppressing this pathway is crucial for obtaining high enantioselectivity. The attempted hydrogenation of methyl-2-acetamidoacrylate identified a deactivation pathway where N–H bond activation generated an iron alkyl κ2-amidate alkyl. For productive catalytic reactions, deuterium labeling studies are consistent with a pathway for hydrogenation involving fast, reversible [2,1]-alkene insertion and a slow, enantiodetermining [1,2]-insertion. Monitoring the catalytic alkene hydrogenation reaction by NMR spectroscopy supports a homogeneous active catalyst that also undergoes C–H activation of the ACNC ligand backbone as a competing reaction. | 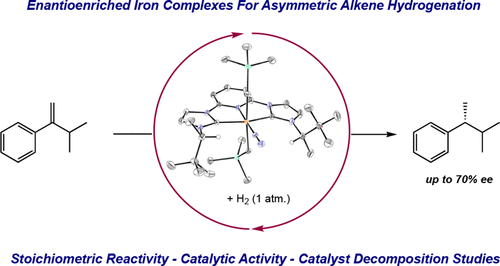 |
217 | A Tutorial on Selectivity Determination in C(sp2)–H Oxidative Addition of Arenes by Transition Metal ComplexesTyler P. Pabst and Paul J. Chirik Organometallics 2021, 40, 813–831. AbstractA Tutorial on factors that determine the selectivity in C(sp2)–H activation and functionalization reactions involving two-electron oxidative addition processes with transition metals is presented. The interplay of the thermodynamics of C(sp2)–H oxidative addition and kinetic influences upon regioselectivity are presented alongside pedagogically valuable experimental and computational results from the literature. Mechanisms and energetics of chelate-assisted C(sp2)–H oxidative addition are examined, as are concepts related to chemoselectivity in the oxidative addition of C(sp2)–H or C(sp2)–X (X = F, Cl, Br, I) bonds with aryl halide substrates. |  |
216 | Iron-catalysed synthesis and chemical recycling of telechelic 1,3-enchained oligocyclobutanesMegan Mohadjer Beromi, C. Rose Kennedy, Jarod M. Younker, Alex Carpenter, Sarah J. Mattler, Joseph A. Throckmorton, and Paul J. Chirik AbstractClosed-loop recycling offers the opportunity to mitigate plastic waste through reversible polymer construction and deconstruction. Although examples of chemical recycling of polymers are known, few have been applied to materials derived from abundant commodity olefinic monomers, which are the building blocks of ubiquitous plastic resins. Here we describe a [2+2] cycloaddition/oligomerization of 1,3-butadiene to yield a previously unrealized telechelic microstructure of (1,n′-divinyl)oligocyclobutane. This material is thermally stable, has stereoregular segments arising from chain-end control, and exhibits high crystallinity even at low molecular weight. Exposure of the oligocyclobutane to vacuum in the presence of the pyridine(diimine) iron precatalyst used to synthesize it resulted in deoligomerization to generate pristine butadiene, demonstrating a rare example of closed-loop chemical recycling of an oligomeric material derived from a commodity hydrocarbon feedstock. |  |
215 | Iron-Catalyzed Vinylsilane Dimerization and Cross-Cycloadditions with 1,3-Dienes: Probing the Origins of Chemo- and RegioselectivityC. Rose Kennedy, Matthew V. Joannou, Janelle E. Steves, Jordan M. Hoyt, Carli B. Kovel, and Paul J. Chirik ACS Catal. 2021, 11, 1368–1379. AbstractThe selective, intermolecular homodimerization and cross-cycloaddition of vinylsilanes with unbiased 1,3-dienes, catalyzed by a pyridine-2,6-diimine (PDI) iron complex are described. In the absence of a diene coupling partner, vinylsilane hydroalkenylation products were obtained chemoselectively with unusual head-to-head regioselectivity (up to >98% purity, 98:2 E/Z). In the presence of a 4- or 2-substituted diene coupling partner, under otherwise identical reaction conditions, formation of value-added [2+2]- and [4+2]-cycloadducts, respectively, was observed. The chemoselectivity profile was distinct from that observed for analogous α-olefin dimerization and cross-reactions with 1,3-dienes. Mechanistic studies conducted with well-defined, single-component precatalysts (MePDI)Fe(L2) (where MePDI = 2,6-(2,6-Me2-C6H3N═CMe)2C5H3N; L2 = butadiene or 2(N2)) provided insights into the kinetic and thermodynamic factors contributing to the substrate-controlled regioselectivity for both the homodimerization and cross-cycloadditions. Diamagnetic iron diene and paramagnetic iron olefin complexes were identified as catalyst resting states, were characterized by in situ nuclear magnetic resonance and Mössbauer spectroscopic studies, and were corroborated with density functional theory calculations. Stoichiometric reactions and computational models provided evidence for a common mechanistic regime where competing steric and electronic requirements dictate the regioselectivity of oxidative cyclization. Although distinct chemoselectivity profiles were observed in cross-cycloadditions with the vinylsilane congeners of α-olefins, these products arose from metallacycles with the same connectivity. The silyl substituents ultimately governed the relative rates of β-H elimination and C–C reductive elimination to dictate final product formation. |  |
214 | Visible-Light-Enhanced Cobalt-Catalyzed Hydrogenations: Switchable Catalysis Enabled by Divergence between Thermal and Photochemical PathwaysLauren N. Mendelsohn, Connor S. MacNeil, Lei Tian, Yoonsu Park, Gregory D. Scholes, and Paul J. Chirik ACS Catal. 2021, 11, 1351–1360. AbstractThe catalytic hydrogenation activity of the readily prepared, coordinatively saturated cobalt(I) precatalyst, (R,R)-(iPrDuPhos)Co(CO)2H ((R,R)-iPrDuPhos = (+)-1,2-bis[(2R,5R)-2,5-diisopropylphospholano]benzene), is described. While efficient turnover was observed with a range of alkenes upon heating to 100 °C, the catalytic performance of the cobalt catalyst was markedly enhanced upon irradiation with blue light at 35 °C. This improved reactivity enabled hydrogenation of terminal, di-, and trisubstituted alkenes, alkynes, and carbonyl compounds. A combination of deuterium labeling studies, hydrogenation of alkenes containing radical clocks, and experiments probing relative rates supports a hydrogen atom transfer pathway under thermal conditions that is enabled by a relatively weak cobalt–hydrogen bond of 54 kcal/mol. In contrast, data for the photocatalytic reactions support light-induced dissociation of a carbonyl ligand followed by a coordination-insertion sequence where the product is released by combination of a cobalt alkyl intermediate with the starting hydride, (R,R)-(iPrDuPhos)Co(CO)2H. These results demonstrate the versatility of catalysis with Earth-abundant metals as pathways involving open- versus closed-shell intermediates can be switched by the energy source. |  |
213 | Mechanistic Origins of Regioselectivity in Cobalt-Catalyzed C(sp2)–H Borylation of Benzoate Esters and Arylboronate EstersTyler P. Pabst, Linda Quach, Kaitlyn T. MacMillan, and Paul J. Chirik AbstractCarbon–hydrogen (C–H) bonds are ubiquitous in organic molecules, and methods for their selective functionalization to more reactive functional groups is a long-standing goal in catalysis, as applied to organic synthesis. Of the established methods involving transition metal catalysts, many employ carefully engineered substrate-catalyst interactions, placing the targeted C–H bond proximal to the metal catalyst, resulting in activation and subsequent functionalization. Here, we report mechanistic investigations describing a conceptual alternative to this approach whereby a cobalt-based borylation catalyst differentiates between subtle electronic differences in C(sp2)-H bonds of benzoate esters and arylboronate esters. These advances motivate studies of catalysts that rely on inherent differences in C–H bond electronics to distinguish chemically inequivalent sites, providing a new tool for organic synthesis. |  |
212 | Ligand Substitution and Electronic Structure Studies of Bis(phosphine)Cobalt Cyclooctadiene Precatalyst for Alkene HydrogenationHongyu Zhong, Megan Mohadjer Beromi, and Paul J. Chirik Can. J. Chem. 2021, 99, 193–201. AbstractDiene self-exchange reactions of the 17-electron, formally cobalt(0) cyclooctadienyl precatalyst, (R,R)-(iPrDuPhos)Co(COD) (P2CoCOD, (R,R)-iPrDuPhos = 1,2-bis((2R,5R)-2,5-diisopropylphospholano)benzene, COD = 1,5-cyclooctadiene) were studied using natural abundance and deuterated 1,5-cyclooctadiene. Exchange of free and coordinated diene was observed at ambient temperature in benzene-d6 solution and kinetic studies support a dissociative process. Both neutral P2CoCOD and the 16-electron, cationic cobalt(I) complex, [(R,R)-(iPrDuPhos)Co(COD)][BArF4] (BArF4 = B[(3,5-(CF3)2)C6H3]4) underwent instantaneous displacement of the 1,5-cyclooctadiene ligand by carbon monoxide and generated the corresponding carbonyl derivatives. The solid-state parameters, DFT-computed Mulliken spin density and analysis of molecular orbitals suggest an alternative description of P2CoCOD as low-spin cobalt(II) with the 1,5-cyclooctadiene acting as a LX2-type ligand. This view of the electronic structure provides insight into the nature of the ligand substitution process and the remarkable stability of the neutral cobalt complexes toward protic solvents observed during catalytic alkene hydrogenation. | 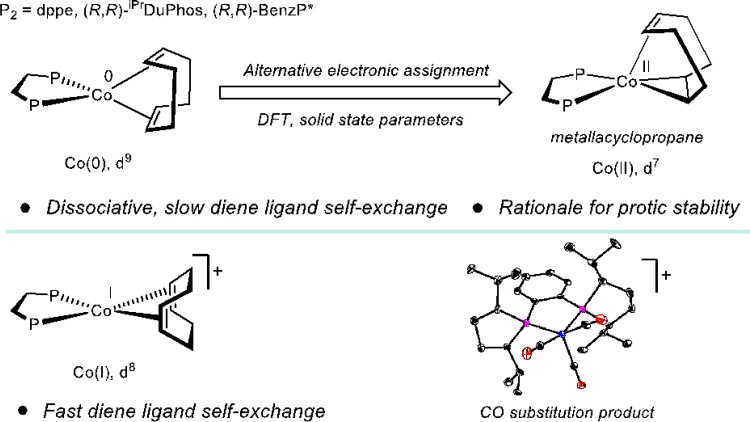 |
211 | Determination of the N–H Bond Dissociation Free Energy in a Pyridine(diimine)molybdenum Complex Prepared by Proton-Coupled Electron TransferGrant W. Margulieux, Sangmin Kim, and Paul J. Chirik Inorg. Chem. 2020, 59, 15394–15401. AbstractThe pyridine(diimine)molybdenum bis(imido) complex (iPrPDI)Mo(═NTol)2 (Tol = 4-methylphenyl) was synthesized by the addition of 2 equiv of 4-methylphenylazide to the corresponding molybdenum benzene derivative, (iPrPDI)Mo(η6-C6H6) [iPrPDI = 2,6-(2,6-iPr2C6H3N═CMe)2C5H3N]. Protonation of (iPrPDI)Mo(═NTol)2 with 2,6-lutinidum triflate yielded a cationic molybdenum amido complex, [(iPrPDI)Mo(NHTol)(═NTol)][OTf], which was further transformed into the neutral molybdenum amido (iPrPDI)Mo(NHTol)(═NTol) by reduction with zinc powder. A series of spectroscopic, synthetic, and pKa determination studies along with electrochemical measurements by the protonation–reduction pathway were used to establish an N–H bond dissociation free energy (BDFE) between 65 and 69 kcal/mol for the molybdenum imido–amido compound, (iPrPDI)Mo(NHTol)(═NTol). Full-molecule density functional theory studies provided a computed value of 61 kcal/mol. By contrast, reduction of (iPrPDI)Mo(═NTol)2 with KC8 afforded the corresponding anionic molybdenum complex K[(iPrPDI)Mo(═NTol)2], which has a potassium cation intercalated with the pyridine and tolyl groups. Protonation of K[(iPrPDI)Mo(═NTol)2] with the weak amidinium acid [TBD(H)][BArF24] (TBD = triazabicyclodecene; BArF24 = B[3,5-(CF3)2C6H3]4) also produced the neutral molybdenum amido complex (iPrPDI)Mo(NHTol)(═NTol). Measurement of the pKa and oxidation potential of K[(iPrPDI)Mo(═NTol)2] provided a range of 69–73 kcal/mol for the N–H BDFE of (iPrPDI)Mo(NHTol)(═NTol), in good agreement with the protonation–reduction route and completing the square scheme. The similar pKa and redox potentials obtained from each pathway demonstrate that both sequences are energetically feasible for proton-coupled electron-transfer (PCET) events. This study on the determination of N–H BDFE of the molybdenum amido complex renders fundamental insight into the N2 reduction cycle by PCET. | 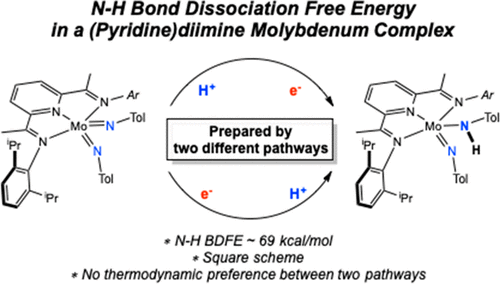 |
210 | Cobalt-Catalyzed C(sp2)–C(sp3) Suzuki–Miyaura Cross CouplingJacob R. Ludwig, Eric M. Simmons, Steven R. Wisniewski, and Paul J. Chirik AbstractA cobalt-catalyzed method for the C(sp2)–C(sp3) Suzuki–Miyaura cross coupling of aryl boronic esters and alkyl bromides is described. Cobalt–ligand combinations were assayed with high-throughput experimentation, and cobalt(II) sources with trans–N,N′-dimethylcyclohexane-1,2-diamine (DMCyDA, L1) produced optimal yield and selectivity. The scope of this transformation encompassed steric and electronic diversity on the aryl boronate nucleophile as well as various levels of branching and synthetically valuable functionality on the electrophile. Radical trap experiments support the formation of electrophile-derived radicals during catalysis. |  |
209 | Coordination-Induced N–H Bond Weakening in a Molybdenum Pyrrolidine Complex: Isotopic Labeling Provides Insight into the Pathway for H2 EvolutionMáté J. Bezdek, István Pelczer, and Paul J. Chirik Organometallics 2020, 39, 3050–3059. AbstractThe synthesis and characterization of a cationic molybdenum pyrrolidine complex are described that exhibits significant coordination-induced N–H bond weakening. The N–H bond dissociation free energy (BDFE) of the coordinated pyrrolidine in [(PhTpy)(PPh2Me)2Mo(NH(pyrr))][BArF24] ([1-NH(pyrr)]+; PhTpy = 4′-Ph-2,2′,6′,2″-terpyridine, NH(pyrr) = pyrrolidine, ArF24 = [C6H3-3,5-(CF3)2]4) was determined to be between 41 and 51 kcal mol–1 by thermochemical analysis and supported by a density functional theory (DFT) computed value of 48 kcal mol–1. The complex [1-NH(pyrr)]+ underwent proton-coupled electron transfer (PCET) to 2,4,6-tri-tert-butylphenoxyl radical, as well as spontaneous H2 evolution upon gentle heating to furnish the corresponding molybdenum pyrrolidide complex [(PhTpy)(PPh2Me)2Mo(N(pyrr))][BArF24] ([1-N(pyrr)]+). Thermolysis of the deuterated isotopologue [1-ND(pyrr)]+ still produced H2 with concomitant incorporation of the isotopic label into the pyrrolidide ligand in the product [(1-N(pyrr–dn)]+ (n = 0–2), consistent with an H2 evolution pathway involving intramolecular H–H bond formation followed by an intermolecular product-forming PCET step. These observations provide the context for understanding H2 evolution in the nonclassical ammine complex [(PhTpy)(PPh2Me)2Mo(NH3)][BArF24] ([1-NH3]+) and are supported by DFT-computed reaction thermochemistry. Overall, these studies offer rare insight into the H2 formation pathway in nonclassical amine complexes with N–H BDFEs below the thermodynamic threshold for H2 evolution and inform the development of well-defined, thermodynamically potent PCET reagents. | 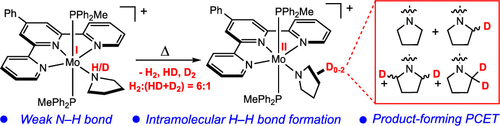 |
208 | Synthesis of Cationic, Dimeric alpha-Diimine Nickel Hydride Complexes and Relevance to the Polymerization of OlefinsNadia G. Léonard, Sam Yruegas, Suzzy C. Ho, Aaron Sattler, Máté J. Bezdek, and Paul J. Chirik Organometallics 2020, 39, 2630–2635. AbstractWell-defined nickel hydride complexes bearing aryl-substituted α-diimine (iPrDI = N,N′-bis(2,6-diisopropylphenyl)-2,3-butanediimine or MesDI = N,N′-bis(2,4,6-trimethylphenyl)-2,3-butanediimine) ligands have been synthesized and studied for the oligomerization of linear internal olefins. Neutral diimine nickel hydride dimers, [(iPrDI)Ni(μ2-H)]2 (1) and [(MesDI)Ni(μ2-H)]2 (2), were explored as synthetic entries to monomeric cationic hydride complexes; however, the dimeric structure was preserved upon oxidation with ferrocenium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate ([Cp2Fe][BArF24]). The monocationic nickel hydride dimers, [(iPrDI)Ni(μ2-H)]2[BArF24] (3) and [(MesDI)Ni(μ2-H)]2[BArF24] (4), were characterized by X-ray diffraction, electron paramagnetic resonance, and nuclear magnetic resonance spectroscopies that support a mixed-valent nickel(I)–nickel(II) assignment. The catalytic activity of 3 and 4 for the oligomerization of 1-hexene, trans-2-hexene, and trans-3-hexene was determined and showed decreased activity as compared to the corresponding nickel dibromide precatalyst activated by methylaluminoxane. |  |
207 | C(sp2)–H Activation with Pyridine Dicarbene Iron Dialkyl Complexes: Hydrogen Isotope Exchange of Arenes Using Benzene-d6 as a Deuterium SourceJavier Corpas, Peter Viereck, and Paul J. Chirik ACS Catal. 2020, 10, 8640–8647. AbstractTreatment of pyridine dicarbene iron dialkyl complexes with low (≪1 atm) pressures of H2 in a benzene-d6 solution promoted rapid hydrogen isotope exchange (HIE) of the C(sp2)–H bonds in both electron-poor and -rich aromatic and heteroaromatic rings with benzene-d6 as the deuterium source. The iron-catalyzed reaction proceeded with predictable regioselectivity, engaging sterically accessible C–H bonds including ortho-to-fluorine sites. The site selectivity for the catalytic HIE reaction was studied to identify the kinetic preferences for C–H activation. Structure–activity relationship studies with a series of iron precatalysts established that introduction of substituents at the 3- and 5-positions of pyridine of the pincer significantly accelerated HIE. Mechanistic studies identified N2 as an inhibitor of C–H activation, while H2 served to generate the active catalyst. |  |
206 | C(sp2)–H Borylation of Heterocycles by Well-Defined Bis(silylene)pyridine Cobalt(III) Precatalysts: Pincer Modification, C(sp2)–H Activation, and Catalytically Relevant IntermediatesRebeca Arévalo, Tyler P. Pabst, and Paul J. Chirik Organometallics 2020, 39, 2763–2773. AbstractWell-defined bis(silylene)pyridine cobalt(III) precatalysts for C(sp2)–H borylation have been synthesized and applied to the investigation of the mechanism of the catalytic borylation of furans and 2,6-lutidine. Specifically, [(ArSiNSi)CoH3]·NaHBEt3 {ArSiNSi = 2,6-[EtNSi(NtBu)2CAr]2C5H3N, where Ar = C6H5 (1-H3·NaHBEt3) or 4-MeC6H4 (2-H3·NaHBEt3)} and trans-[(ArSiNSi)Co(H)2BPin] {Ar = C6H5 [1-(H)2BPin] or 4-MeC6H4 [2-(H)2BPin], and Pin = pinacolato} were prepared and employed as single-component precatalysts for the C(sp2)–H borylation of 2-methylfuran, benzofuran, and 2,6-lutidine. The cobalt(III) precursors, 2-H3·NaHBEt3 and 2-(H)2BPin, also promoted C(sp2)–H activation of benzofuran, yielding [(ArSiNSi)CoH(Bf)2] {Ar = 4-MeC6H4 [2-H(Bf)2], and Bf = 2-benzofuranyl}. Monitoring the catalytic borylation of 2-methylfuran and 2,6-lutidine by 1H NMR spectroscopy established the trans-dihydride cobalt(III) boryl as the catalyst resting state at low substrate conversions. At higher conversions, two distinct pincer modification pathways were identified, depending on the substrate and the boron source. |  |
205 | Beyond Ammonia: Nitrogen–Element Bond Forming Reactions with Coordinated DinitrogenSangmin Kim, Florian Loose, and Paul J. Chirik Chem. Rev. 2020, 120, 5637–5681. AbstractThe functionalization of coordinated dinitrogen to form nitrogen–element bonds en route to nitrogen-containing molecules is a long-standing challenge in chemical synthesis. The strong triple bond and the nonpolarity of the N2 molecule pose thermodynamic and kinetic challenges for promoting reactivity. While heterogeneous, homogeneous, and biological catalysts are all known for catalytic nitrogen fixation to ammonia, the catalytic synthesis of more complicated nitrogen-containing organic molecules has far less precedent. The example of silyl radical additions to coordinated nitrogen to form silylamines stands as the lone example of a catalytic reaction involving N2 to form a product other than ammonia. This Review surveys the field of molecular transition metal complexes as well as recent boron examples for the formation of nitrogen–element bonds. Emphasis is placed on the coordination and activation modes of N2 in the various metal compounds from across the transition series and how these structures can rationally inform reactivity studies. Over the past few decades, the field has evolved from the addition of carbon electrophiles in a manner similar to that of protonation reactions to more organometallic-inspired reactivity, including insertions, 1,2-additions, and cycloadditions. Various N–C, N–Si, and N–B bond-forming reactions have been discovered, highlighting that the challenge for catalytic chemistry is not in the reactivity of coordinated dinitrogen but rather removal of the functionalized ligand from the coordination sphere of the metal. |  |
204 | Catalytic Hydrogenation of a Manganese(V) Nitride to AmmoniaSangmin Kim, Hongyu Zhong, Yoonsu Park, Florian Loose, and Paul J. Chirik J. Am. Chem. Soc. 2020, 142, 9518–9524. AbstractThe catalytic hydrogenation of a metal nitride to produce free ammonia using a rhodium hydride catalyst that promotes H2 activation and hydrogen-atom transfer is described. The phenylimine-substituted rhodium complex (η5-C5Me5)Rh(MePhI)H (MePhI = N-methyl-1-phenylethan-1-imine) exhibited higher thermal stability compared to the previously reported (η5-C5Me5)Rh(ppy)H (ppy = 2-phenylpyridine). DFT calculations established that the two rhodium complexes have comparable Rh–H bond dissociation free energies of 51.8 kcal mol–1 for (η5-C5Me5)Rh(MePhI)H and 51.1 kcal mol–1 for (η5-C5Me5)Rh(ppy)H. In the presence of 10 mol% of the phenylimine rhodium precatalyst and 4 atm of H2 in THF, the manganese nitride (tBuSalen)Mn≡N underwent hydrogenation to liberate free ammonia with up to 6 total turnovers of NH3 or 18 turnovers of H• transfer. The phenylpyridine analogue proved inactive for ammonia synthesis under identical conditions owing to competing deleterious hydride transfer chemistry. Subsequent studies showed that the use of a non-polar solvent such as benzene suppressed formation of the cationic rhodium product resulting from the hydride transfer and enabled catalytic ammonia synthesis by proton-coupled electron transfer. |  |
203 | Iron Catalyzed Synthesis and Chemical Recycling of Telechelic, 1,3-Enchained OligocyclobutanesMegan Mohadjer Beromi, C. Rose Kennedy, Jarod M. Younker, Alex E. Carpenter, Sarah J. Mattler, Joseph A. Throckmorton, and Paul J. Chirik AbstractClosed-loop recycling offers the opportunity to help mitigate plastic waste through reversible polymer construction and deconstruction. While examples of the chemical recycling polymers are known, few have been applied to materials derived from abundant commodity olefinic monomers that are the building blocks of ubiquitous plastic resins. Here we describe a [2+2] cycloaddition oligomerization of 1,3-butadiene to yield a previously unrealized telechelic microstructure of (1,n’-divinyl)oligocyclobutane. This material is thermally stable, has stereoregular segments arising from chain-end control, and exhibits high crystallinity even at low molecular weight. Exposure of the oligocyclobutane to vacuum in the presence of the pyridine(diimine) iron precatalyst used to synthesize it resulted in deoligomerization to generate pristine butadiene, demonstrating a rare example of closed-loop chemical recycling of an oligomeric material derived from a commodity hydrocarbon feedstock. |  |
202 | Synthesis and Reactivity of Organometallic Intermediates Relevant to Cobalt-Catalyzed HydroformylationConnor S. MacNeil, Lauren N. Mendelsohn, Hongyu Zhong, Tyler P. Pabst, and Paul J. Chirik Angew. Chem. Int. Ed. 2020, 59, 8912–8916. AbstractIntermediates relevant to cobalt-catalyzed alkene hydroformylation have been isolated and evaluated in fundamental organometallic transformations relevant to aldehyde formation. The 18-electron (R,R)-(iPrDuPhos)Co(CO)2H has been structurally characterized, and it promotes exclusive hydrogenation of styrene in the presence of 50 bar of H2/CO gas (1:1) at 100 °C. Deuterium-labeling studies established reversible 2,1-insertion of styrene into the Co−D bond of (R,R)-(iPrDuPhos)Co(CO)2D. Whereas rapid β-hydrogen elimination from cobalt alkyls occurred under an N2 atmosphere, alkylation of (R,R)-(iPrDuPhos)Co(CO)2Cl in the presence of CO enabled the interception of (R,R)-(iPrDuPhos)Co(CO)2C(O)CH2CH2Ph, which upon hydrogenolysis under 4 atm H2 produced the corresponding aldehyde and cobalt hydride, demonstrating the feasibility of elementary steps in hydroformylation. Both the hydride and chloride derivatives, (X=H−, Cl−), underwent exchange with free 13CO. Under reduced pressure, (R,R)-(iPrDuPhos)Co(CO)2Cl underwent CO dissociation to form (R,R)-(iPrDuPhos)Co(CO)Cl. |  |
201 | Investigations into the Mechanism of Inter- and Intramolecular Iron-Catalyzed [2+2] Cycloaddition of AlkenesMatthew V. Joannou, Jordan M. Hoyt, and Paul J. Chirik J. Am. Chem. Soc. 2020, 142, 5314–5330. AbstractMechanistic studies are reported on the inter- and intramolecular [2 + 2] alkene cycloadditions to form cyclobutanes promoted by (tricPDI)Fe(N2) (tricPDI = 2,6-(2,4,6-tricyclopentyl)C6H2N = CMe)2C5H3N). A combination of kinetic measurements, freeze-quench 57Fe Mössbauer and infrared spectroscopic measurements, deuterium labeling studies, natural abundance 13C KIE studies, and isolation and characterization of catalytically relevant intermediates were used to gain insight into the mechanism of both inter- and intramolecular [2 + 2] cycloaddition reactions. For the stereo- and regioselective [2 + 2] cycloaddition of 1-octene to form trans-1,2-dihexylcyclobutane, a first-order dependence on both iron complex and alkene was measured as well as an inverse dependence on N2 pressure. Both 57Fe Mössbauer and infrared spectroscopic measurements identified (tricPDI)Fe(N2)(η2-1-octene) as the catalyst resting state. Rate-determining association of 1-octene to (tricPDI)Fe(η2-1-octene) accounts for the first order dependence of alkene and the inverse dependence on N2. Heavy atom 13C/12C kinetic isotope effects near unity also support post rate-determining C–C bond formation. By contrast, the intramolecular iron-catalyzed [2 + 2] cycloaddition of 1,7-octadiene yielded cis-bicyclo[4.2.0]octane in 92:8 d.r. and a first order dependence on the iron precursor and zeroth order behavior in both diene and N2 pressure were measured. A pyridine(diimine) iron trans-bimetallacycle was identified as the catalyst resting state and was isolated and characterized by X-ray diffraction and 1H NMR and 57Fe Mössbauer spectroscopies. Dissolution of the iron trans-bimetallacycle in benzene-d6 produced predominantly the cis-cyclobutane product, establishing interconversion between the trans and cis metallacycles during the catalytic reaction and consistent with a Curtin-Hammett kinetic regime. A primary 13C/12C kinetic isotope effect of 1.022(4) was measured at 23 °C, consistent with irreversible unimolecular reductive elimination to form the cyclobutane product. Despite complications from competing cyclometalation of chelate aryl substituents, deuterium labeling experiments were consistent with unimolecular C–C reductive elimination that occurred either by a concerted pathway or a radical rebound sequence that is faster than C–C bond rotation. | ![Investigations into the Mechanism of Inter- and Intramolecular Iron-Catalyzed [2+2] Cycloaddition of Alkenes](/wp-content/uploads/2022/10/img-publ-201.gif) |
200 | Cobalt-Catalyzed Asymmetric Hydrogenation of alpha, beta-Unsaturated Carboxylic Acids by Homolytic H2 CleavageHongyu Zhong, Michael Shevlin, and Paul J. Chirik J. Am. Chem. Soc. 2020, 142, 5272–5281. AbstractThe asymmetric hydrogenation of α,β-unsaturated carboxylic acids using readily prepared bis(phosphine) cobalt(0) 1,5-cyclooctadiene precatalysts is described. Di-, tri-, and tetra-substituted acrylic acid derivatives with various substitution patterns as well as dehydro-α-amino acid derivatives were hydrogenated with high yields and enantioselectivities, affording chiral carboxylic acids including Naproxen, (S)-Flurbiprofen, and a d-DOPA precursor. Turnover numbers of up to 200 were routinely obtained. Compatibility with common organic functional groups was observed with the reduced cobalt(0) precatalysts, and protic solvents such as methanol and isopropanol were identified as optimal. A series of bis(phosphine) cobalt(II) bis(pivalate) complexes, which bear structural similarity to state-of-the-art ruthenium(II) catalysts, were synthesized, characterized, and proved catalytically competent. X-band EPR experiments revealed bis(phosphine)cobalt(II) bis(carboxylate)s were generated in catalytic reactions and were identified as catalyst resting states. Isolation and characterization of a cobalt(II)–substrate complex from a stoichiometric reaction suggests that alkene insertion into the cobalt hydride occurred in the presence of free carboxylic acid, producing the same alkane enantiomer as that from the catalytic reaction. Deuterium labeling studies established homolytic H2 (or D2) activation by Co(0) and cis addition of H2 (or D2) across alkene double bonds, reminiscent of rhodium(I) catalysts but distinct from ruthenium(II) and nickel(II) carboxylates that operate by heterolytic H2 cleavage pathways. |  |
199 | A Boron Activating Effect Enables Cobalt-Catalyzed Asymmetric Hydrogenation of Sterically Hindered AlkenesPeter Viereck, Simon Krautwald, Tyler P. Pabst and Paul J. Chirik J. Am. Chem. Soc. 2020, 142, 3923–3930. AbstractUnsymmetric 1,1-diboryl alkenes bearing one −[BPin] (BPin = pinacolatoboryl) and one −[BDan] (BDan = 1,8-diaminonaphthalatoboryl) substituent each were hydrogenated in high yield and enantioselectivity using C1-symmetric pyridine(diimine) (PDI) cobalt complexes. High activities and stereoselectivities were observed with an array of 2-alkyl-, 2-aryl-, and 2-boryl-substituted 1,1-diboryl alkenes, giving rise to enantioenriched diborylalkane building blocks. Systematic study of substrate substituent effects identified competing steric and electronic demands in the key activating role of the boron substituents, whereby sterically unencumbered boronates such as −[BDan], −[BCat] (BCat = catecholatoboryl), and −[Beg] (Beg = ethylene glycolatoboryl) promote the hydrogenation of trisubstituted alkenes by enabling irreversible α-boron-directed insertion pathways to achieve otherwise challenging hydrogenations of trisubstituted alkenes. Deuterium-labeling studies with 1,1-diboryl alkenes support an insertion pathway generating a chiral intermediate with two different boron substituents and cobalt bound to the same carbon. |  |
198 | Ketone Synthesis from Benzyldiboronates and Esters: Leveraging alpha-Boryl Carbanions for Carbon–Carbon Bond FormationBoran Lee and Paul J. Chirik J. Am. Chem. Soc. 2020, 142, 2429–2437. AbstractAn alkoxide-promoted method for the synthesis of ketones from readily available esters and benzyldiboronates is described. The synthetic method is compatible with a host of sterically differentiated alkyl groups, alkenes, acidic protons α to carbonyl groups, tertiary amides, and aryl rings having common organic functional groups. With esters bearing α-stereocenters, high enantiomeric excess was maintained during ketone formation, establishing minimal competing racemization by deprotonation. Monitoring the reaction between benzyldiboronate and LiOtBu in THF at 23 °C allowed for the identification of products arising from deborylation to form an α-boryl carbanion, deprotonation, and alkoxide addition to form an “-ate” complex. Addition of 4-trifluoromethylbenzoate to this mixture established the α-boryl carbanion as the intermediate responsible for C–C bond formation and ultimately ketone synthesis. Elucidation of the role of this intermediate leveraged additional bond-forming chemistry and enabled the one-pot synthesis of ketones with α-halogen atoms and quaternary centers with four-different carbon substituents. |  |
197 | Direct Observation of Transmetalation from a Neutral Boronate Ester to a Pyridine(diimine) Iron AlkoxidePaul Peterson, Stephan Rummelt, Bradley Wile, S. Chantal Stieber, Hongyu Zhong and Paul J. Chirik Organometallics 2020, 1, 201–205. AbstractTransmetalation of the neutral boronate esters, (2-benzofuranyl)BPin and (2-benzofuranyl)BNeo (Pin = pinacolato, Neo = neopentylglycolato), to a representative pyridine(diimine) iron alkoxide complex, (iPrPDI)FeOEt (iPrPDI = 2,6-(2,6-iPr2–C6H3N═CMe)2C5H3N; R = Me, Et, SiMe3), to yield the corresponding iron benzofuranyl derivative was studied. Synthesis of the requisite iron alkoxide complexes was accomplished either by salt metathesis between (iPrPDI)FeCl and NaOR (R = Me, Et, SiMe3) or by protonation of the iron alkyl, (iPrPDI)FeCH2SiMe3, by the free alcohol R′OH (R′ = Me, Et). A combination of magnetic measurements, X-ray diffraction, NMR, and Mössbauer spectroscopies and DFT calculations identified each (iPrPDI)FeOR compound as an essentially planar, high-spin, S = 3/2 compound where the iron is engaged in antiferromagnetic coupling with a radical anion on the chelate (STotal = 3/2; SFe = 2, SPDI = −1/2). The resulting iron benzofuranyl product, (iPrPDI)Fe(2-benzofuranyl), was characterized by X-ray diffraction and in combination with magnetic measurements, spectroscopic and computational data, was identified as an overall S = 1/2 compound, demonstrating that a net spin-state change accompanies transmetalation (SFe = 1, SPDI = −1/2). These findings may be relevant to further development of iron-catalyzed Suzuki–Miyaura cross-coupling with neutral boronate esters and alkoxide bases. |  |
196 | Pyridine(diimine) Iron Diene Complexes Relevant to Catalytic [2+2]-Cycloaddition ReactionsC. Rose Kennedy, Hongyu Zhong, Matthew Joannou, and Paul J. Chirik Adv. Syn. Cat. 2020, 2, 404–416. AbstractThe synthesis, characterization, and catalytic activity of pyridine(diimine) iron piperylene and isoprene complexes are described. These diene complexes are competent precatalysts for (i) the selective cross-[2+2]-cycloaddition of butadiene or (E)-piperylene with ethylene and α-olefins and (ii) the 1,4-hydrovinylation of isoprene with ethylene. In the former case, kinetic analysis implicated the diamagnetic η4-piperylene complex as the resting state prior to rate-determining oxidative cyclization. Variable temperature 1H NMR and EXSY experiments established that diene exchange from the diamagnetic, 18e– complexes occurred rapidly in solution at ambient temperature through a dissociative mechanism. The solid-state structure of Me(Et)PDI Fe(η4-piperylene) (Me(Et)PDI=2,6-(2,6-Me2-C6H3N=CEt)2C5H3N) was determined by single-crystal X-ray diffraction and confirmed the s-trans coordination of the monosubstituted 1,3-diene. Possible relationships between ligand-controlled diene coordination geometry, metallacycle denticity, and chemoselectivity of iron-mediated cycloaddition reactions are discussed. | ![Pyridine(diimine) Iron Diene Complexes Relevant to Catalytic [2+2]-Cycloaddition Reactions](/wp-content/uploads/2023/04/img-publ-196.webp) |
195 | Hydrogenation of N-Heteroarenes Using Rhodium Precatalysts: Reductive Elimination Leads to Formation of Multimetallic ClustersSangmin Kim, Florian Loose, Máté J. Bezdek, Xiaoping Wang, and Paul J. Chirik J. Am. Chem. Soc. 2019, 141, 17900–17908. AbstractA rhodium-catalyzed method for the hydrogenation of N-heteroarenes is described. A diverse array of unsubstituted N-heteroarenes including pyridine, pyrrole, and pyrazine, traditionally challenging substrates for hydrogenation, were successfully hydrogenated using the organometallic precatalysts, [(η5-C5Me5)Rh(N-C)H] (N-C = 2-phenylpyridinyl (ppy) or benzo[h]quinolinyl (bq)). In addition, the hydrogenation of polyaromatic N-heteroarenes exhibited uncommon chemoselectivity. Studies into catalyst activation revealed that photochemical or thermal activation of [(η5-C5Me5)Rh(bq)H] induced C(sp2)–H reductive elimination and generated the bimetallic complex, [(η5-C5Me5)Rh(μ2,η2-bq)Rh(η5-C5Me5)H]. In the presence of H2, both of the [(η5-C5Me5)Rh(N-C)H] precursors and [(η5-C5Me5)Rh(μ2,η2-bq)Rh(η5-C5Me5)H] converted to a pentametallic rhodium hydride cluster, [(η5-C5Me5)4Rh5H7], the structure of which was established by NMR spectroscopy, X-ray diffraction, and neutron diffraction. Kinetic studies on pyridine hydrogenation were conducted with each of the isolated rhodium complexes to identify catalytically relevant species. The data are most consistent with hydrogenation catalysis prompted by an unobserved multimetallic cluster with formation of [(η5-C5Me5)4Rh5H7] serving as a deactivation pathway. | 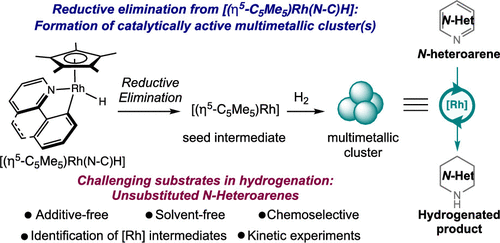 |
194 | [4+4]-Cycloaddition of Isoprene for the production of High-Performance Bio-Based Jet FuelKyle Rosenkoetter, C. Rose Kennedy, Paul Chirik, and Benjamin Grant Harvey Green Chem. 2019, 21, 5616–5623. AbstractIsoprene was efficiently converted to 1,6-dimethyl-1,5-cyclooctadiene (DMCOD) by selective [4 + 4]-cycloaddition with a catalyst formed by in situ reduction of [(MePI)FeCl(μ-Cl)]2 (MePI = [2-(2,6-(CH3)2-C6H3-N | ![[4+4]-Cycloaddition of Isoprene for the production of High-Performance Bio-Based Jet Fuel](/wp-content/uploads/2022/10/img-publ-194.gif) |
193 | Cobalt-Catalyzed Borylation of Fluorinated Arenes: Thermodynamic Control of C(sp2)–H Oxidative Addition Results in ortho-to-Fluorine SelectivityTyler P. Pabst, Jennifer V. Obligacion, Étienne Rochette, Iraklis Pappas, and Paul J. Chirik J. Am. Chem. Soc. 2019, 141, 15378–15389. AbstractThe mechanism of C(sp2)–H borylation of fluorinated arenes with B2Pin2 (Pin = pinacolato) catalyzed by bis(phosphino)pyridine (iPrPNP) cobalt complexes was studied to understand the origins of the uniquely high ortho-to-fluorine regioselectivity observed in these reactions. Variable time normalization analysis (VTNA) of reaction time courses and deuterium kinetic isotope effect measurements established a kinetic regime wherein C(sp2)–H oxidative addition is fast and reversible. Monitoring the reaction by in situ NMR spectroscopy revealed the intermediacy of a cobalt(I)–aryl complex that was generated with the same high ortho-to-fluorine regioselectivity associated with the overall catalytic transformation. Deuterium labeling experiments and stoichiometric studies established C(sp2)–H oxidative addition of the fluorinated arene as the selectivity-determining step of the reaction. This step favors the formation of ortho-fluoroaryl cobalt intermediates due to the ortho fluorine effect, a phenomenon whereby ortho fluorine substituents stabilize transition metal–carbon bonds. Computational studies provided evidence that the cobalt–carbon bonds of the relevant intermediates in (iPrPNP)Co-catalyzed borylation are strengthened with increasing ortho fluorine substitution. The atypical kinetic regime involving fast and reversible C(sp2)–H oxidative addition in combination with the thermodynamic preference for forming cobalt–aryl bonds adjacent to fluorinated sites are the origin of the high regioselectivity in the catalytic borylation reaction. | 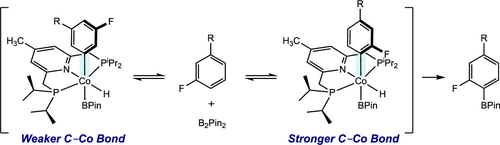 |
192 | Remote, Diastereoselective Cobalt-Catalyzed Alkene Isomerization-Hydroboration: Access to Stereodefined 1,3-Difunctionalized IndanesNadia G. Léonard, W. Neil Palmer, Max R. Friedfeld, Máté J. Bezdek, and Paul J. Chirik ACS Catal. 2019, 9, 9034–9044. AbstractThe remote, diastereoselective hydroboration of 2- and 3-substituted indenes with a 2,2′:6′,2″-terpyridine cobalt alkyl precatalyst is described that maintains high regio- and stereoselectivity independent of the starting position of the alkene. Several 1,2- and 1,3-disubstituted indanyl boronate esters were obtained with exclusive (>20:1 dr) selectivity for the trans diastereomer including synthetically versatile, stereodefined diboron derivatives. Alkene isomerization by a putative cobalt hydride intermediate precedes carbon–boron bond formation, leading to the observed regioselectivity for boron incorporation at the unsubstituted C(sp3)–H benzylic site. The regio- and diastereoselectivity of the transformation were maintained independent of the starting position of the alkene, as demonstrated by hydroboration of three isomers of methyl-substituted indene. Deuterium-labeling experiments support rapid and reversible insertion and β-hydride elimination to isomerize 3-methylindene and 1-exo-methylene-indane, accounting for the isotopic distribution observed in the products. Mechanistic studies, including stoichiometric experiments, density functional theory calculations, and kinetic analysis, support a mechanism in which 2,3-alkene insertion into a cobalt hydride intermediate determines both the regio- and diastereoselectivity of the catalytic reaction. Synthetic applications of the indanyl boronate esters were demonstrated through the elaboration of the products to several examples of 1,3-disubstituted indanes, important carbocyclic structural motifs in both pharmacological and bioactive molecules. |  |
191 | Synthesis, Structure, and Hydrogenolysis of Pyridine Dicarbene Iron Dialkyl ComplexesStephan M. Rummelt, Jonathan M. Darmon, Renyuan Pony Yu, Peter Viereck, Tyler P. Pabst, Zoë R. Turner, Grant W. Margulieux, Shunlin Gu, and Paul J. Chirik Organometallics 2019, 38, 3159–3168. AbstractTwo methods for the synthesis of bis(imidazol-2-ylidene)pyridine iron dialkyl complexes, (CNC)Fe(CH2SiMe3)2, have been developed. The first route consists of addition of 2 equiv of LiCH2SiMe3 to the iron dihalide complex (CNC)FeBr2, while the second relies on addition of the free CNC ligand to the readily prepared (py)2Fe(CH2SiMe3)2 (py = pyridine). With aryl-substituted CNC ligands, octahedral complexes of the type (ArCNC)Fe(CH2SiMe3)2(N2) (ArCNC = bis(arylimidazol-2-ylidene)pyridine) were isolated, where the dinitrogen ligand occupies the site trans to the pyridine of the CNC chelate. In contrast, the alkyl-substituted variant (tBuACNC)Fe(CH2SiMe3)2 (tBuACNC = 2,6-(tBu-imidazol-2-ylidene)2pyridine) was isolated as the five-coordinate compound lacking dinitrogen. Exposure of the (ArCNC)Fe(CH2SiMe3)2(N2) derivatives to an H2 atmosphere resulted in formation of the corresponding iron hydride complexes (ArCNC)FeH4. These compounds catalyzed hydrogen isotope exchange between the deuterated benzene solvent and H2, generating isotopologues and isotopomers of (ArCNC)Fe(Hn)(D4–n) (n = 0–4). When (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) (3,5-Me2MesCNC = 2,6-(2,4,6-Me3-C6H2-imidazol-2-ylidene)2-3,5-Me2-pyridine) was treated successively with H2 and then N2, the corresponding reduced dinitrogen complex (3,5-Me2MesCNC)Fe(N2)2 was isolated. The same product was also obtained following addition of pinacolborane to (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2). |  |
190 | Enabling Two-Electron Pathways with Iron and Cobalt: From Ligand Design to Catalytic ApplicationsRebeca Arévalo and Paul J. Chirik J. Am. Chem. Soc. 2019, 141, 9106–9123. AbstractHomogeneous catalysis with Earth-abundant, first-row transition metals, including iron and cobalt, has gained considerable recent attention as a potentially cost-effective and sustainable alternative to more commonly and historically used precious metals. Because fundamental organometallic transformations, such as oxidative addition and reductive elimination, are two-electron processes and essential steps in many important catalytic cycles, controlling redox chemistry—in particular overcoming one-electron chemistry—has been as a central challenge with Earth-abundant metals. This Perspective focuses on approaches to impart sufficiently strong ligand fields to generate electron-rich metal complexes able to promote oxidative addition reactions where the redox changes are exclusively metal-based. Emphasis is placed on how ligand design and exploration of fundamental organometallic chemistry coupled with mechanistic understanding have been used to discover iron catalysts for the hydrogen isotope exchange in pharmaceuticals and cobalt catalysts for C(sp2)–H borylation reactions. A pervasive theme is that first-row metal complexes often promote unique chemistry from their precious-metal counterparts, demonstrating that these elements offer a host of new opportunities for reaction discovery and for more sustainable catalysis. |  |
189 | Syntheses and Catalytic Hydrogenation Performance of Cationic Bis(phosphine) Cobalt(I) Diene and Arene CompoundsHongyu Zhong, Max R. Friedfeld, and Paul J. Chirik Angew. Chem. Int. Ed. 2019, 58, 9194–9198. AbstractChloride abstraction from [(R,R)-(iPrDuPhos)Co(μ-Cl)]2 with NaBArF4 (BArF4=B[(3,5-(CF3)2)C6H3]4) in the presence of dienes, such as 1,5-cyclooctadiene (COD) or norbornadiene (NBD), yielded long sought-after cationic bis(phosphine) cobalt complexes, [(R,R)-(iPrDuPhos)Co(η2,η2-diene)][BArF4]. The COD complex proved substitutionally labile undergoing diene substitution with tetrahydrofuran, NBD, or arenes. The resulting 18-electron, cationic cobalt(I) arene complexes, as well as the [(R,R)-(iPrDuPhos)Co(diene)][BArF4] derivatives, proved to be highly active and enantioselective precatalysts for asymmetric alkene hydrogenation. A cobalt–substrate complex, [(R,R)-(iPrDuPhos)Co(MAA)][BArF4] (MAA=methyl 2-acetamidoacrylate) was crystallographically characterized as the opposite diastereomer to that expected for productive hydrogenation demonstrating a Curtin–Hammett kinetic regime similar to rhodium catalysis. |  |
188 | Regio- and Diastereoselective Iron-Catalyzed [4+4]-Cycloaddition of 1,3-DienesC. Rose Kennedy, Hongyu Zhong, Rachel L. Macaulay, and Paul J. Chirik J. Am. Chem. Soc. 2019, 141, 8557–8573. AbstractA family of single-component iron precatalysts for the [4+4]-cyclodimerization and intermolecular cross-[4+4]-cycloaddition of monosubstituted 1,3-dienes is described. Cyclooctadiene products were obtained with high regioselectivity, and catalyst-controlled access to either cis– or trans-diastereomers was achieved using 4-substituted diene substrates. Reactions conducted either with single-component precatalysts or with iron dihalide complexes activated in situ proved compatible with common organic functional groups and were applied on multigram scale (up to >100 g). Catalytically relevant, S = 1 iron complexes bearing 2-(imino)pyridine ligands, (RPI)FeL2 (RPI = [2-(2,6-R2-C6H3-N═CMe)-C5H4N] where R = iPr or Me, L2 = bis-olefin), were characterized by single-crystal X-ray diffraction, Mößbauer spectroscopy, magnetic measurements, and DFT calculations. The structural and spectroscopic parameters are consistent with an electronic structure description comprised of a high spin iron(I) center (SFe = 3/2) engaged in antiferromagnetically coupling with a ligand radical anion (SPI = −1/2). Mechanistic studies conducted with these single-component precatalysts, including kinetic analyses, 12C/13C isotope effect measurements, and in situ Mößbauer spectroscopy, support a mechanism involving oxidative cyclization of two dienes that determines regio- and diastereoselectivity. Topographic steric maps derived from crystallographic data provided insights into the basis for the catalyst control through stereoselective oxidative cyclization and subsequent, stereospecific allyl-isomerization and C–C bond-forming reductive elimination. | ![Regio- and Diastereoselective Iron-Catalyzed [4+4]-Cycloaddition of 1,3-Dienes](/wp-content/uploads/2022/10/img-publ-188.png) |
187 | Evaluation of excited state bond weakening for ammonia synthesis from a manganese nitride: stepwise proton coupled electron transfer is preferred over hydrogen atom transferFlorian Loose, Dian Wang, Lei Tian, Gregory D. Scholes, Robert R. Knowles and Paul J. Chirik Chem. Comm. 2019, 55, 5595–5598. AbstractConcepts for the thermodynamically challenging synthesis of weak N–H bonds by photoinduced proton coupled electron transfer are explored. Upon irradiation with blue light, ammonia synthesis was achieved from the manganese nitride (tBuSalen)MnN (tBuSalen = (S,S)-(+)-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamine) in the presence of 9,10-dihydroacridine and a ruthenium photocatalyst in iPrOH solution. Although in one case the ruthenium complex bears a remote N–H bond that weakens to 41 kcal mol−1 upon irradiation, control experiments with the N-methylated analog demonstrate the ruthenium complex serves as a photoreductant rather than hydrogen-atom transfer catalyst in aprotic solvents. Luminescence quenching experiments support a ruthenium(II)/(III) cycle rather than a ruthenium(I)/(II) alternative. Identification of the manganese complex following ammonia synthesis was also accomplished. |  |
186 | A fresh approach to synthesizing ammonia from air and waterMáté J. Bezdek and Paul J. Chirik AbstractGlobal food production requires ammonia-based fertilizers. The industrial transformation of atmospheric nitrogen gas (N2, also known as dinitrogen) into ammonia (NH3) is therefore essential for human life. Despite the simplicity of the molecules involved, the cleavage of the strong nitrogen–nitrogen triple bond (the N≡N bond) in dinitrogen and the concomitant formation of nitrogen–hydrogen (N–H) bonds poses a difficult challenge for catalytic chemistry, and typically involves conditions that are costly in terms of energy requirements: high reaction temperatures, high pressures or combinations of reactive reagents that are difficult to handle and energy-intensive to make. Writing in Nature 2019, 568, 536–540, Ashida, Y., Arashiba, K., Nakajima, K. & Nishibayashi, Y. demonstrate that a samarium compound mixed with water and combined with a molybdenum catalyst can promote ammonia synthesis from dinitrogen under ambient conditions. The work opens up avenues of research in the hunt for ammonia-making processes that operate under ambient conditions, and raises the question of what an ideal process should be. |  |
185 | Pyridine(diimine) Chelate Hydrogenation in a Molybdenum Nitrido Ethylene ComplexMáté J. Bezdek and Paul J. Chirik Organometallics 2019, 38, 1682–1687. AbstractAddition of H2 gas to the aryl-substituted pyridine(diimine) molybdenum nitride (iPrPDI)Mo(N)(C2H4) ([1-(N)(η2-C2H4)]; iPrPDI = 2,6-(2,6-iPr2-C6H3N═CMe)2C5H3N) in the presence of the rhodium hydride precatalyst (η5-C5Me5)(py-Ph)Rh(H) ([Rh–H]; py-Ph = 2-phenylpyridine) resulted in partial hydrogenation of the central pyridine of the iPrPDI chelate to yield (iPrTHPDI)Mo(N)(C2H4) ([2-(N)(η2-C2H4)]; iPrTHPDI = 2,6-(2,6-iPr2-C6H3N═CMe)2C5H6N). The product, [2-(N)(η2-C2H4)], was structurally and spectroscopically characterized, and its electronic structure was examined by density functional theory (DFT). The stepwise addition of H atoms from [Rh–H] to [1-(N)(η2-C2H4)] was computed to be thermodynamically viable by DFT. | 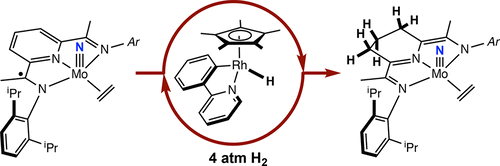 |
184 | Ni(I)–X Complexes Bearing a Bulky alpha-Diimine Ligand: Synthesis, Structure and Superior Catalytic Performance in the Hydrogen Isotope Exchange of PharmaceuticalsCayetana Zarate, Haifeng Yang, Máté J. Bezdek, David Hesk and Paul J. Chirik J. Am. Chem. Soc. 2019, 141, 5034–5044. AbstractThe synthesis and spectroscopic characterization of a family of Ni–X (X = Cl, Br, I, H) complexes supported by the bulky α-diimine chelate N,N′-bis(1R,2R,3R,5S)-(−)-isopinocampheyl-2,3-butanediimine (ipcADI) are described. Diimine-supported, three-coordinate nickel(I)–X complexes have been proposed as key intermediates in a host of catalytic transformations such as C–C and C–heteroatom cross-coupling and C–H functionalization but have until now remained synthetically elusive. A combination of structural, spectroscopic, electrochemical, and computational studies were used to establish the electronic structure of each monomeric [(ipcADI)NiX] (X = Cl, Br, I) complex as a nickel(I) derivative supported by a redox-neutral α-diimine chelate. The dimeric nickel hydride, [(ipcADI)Ni(μ2-H)]2, was prepared and characterized by X-ray diffraction; however, magnetic measurements and 1H NMR spectroscopy support monomer formation at ambient temperature in THF solution. This nickel hydride was used as a precatalyst for the hydrogen isotope exchange (HIE) of C–H bonds in arenes and pharmaceuticals. By virtue of the multisite reactivity and high efficiency, the new nickel precatalyst provided unprecedented high specific activities (50–99 Ci/mmol) in radiolabeling, meeting the threshold required for radioligand binding assays. Use of air-stable and readily synthesized nickel precursor, [(ipcADI)NiBr2], broad functional group tolerance, and compatibility with polar protic solvents are additional assets of the nickel-catalyzed HIE method. |  |
183 | N–H Bond Formation in a Manganese(V) Nitride Yields Ammonia by Light-Driven Proton-Coupled Electron TransferDian Wang, Florian Loose, Paul J. Chirik, and Robert R. Knowles J. Am. Chem. Soc. 2019, 141, 4795–4799. AbstractA method for the reduction of a manganese nitride to ammonia is reported, where light-driven proton-coupled electron transfer enables the formation of weak N—H bonds. Photoreduction of (saltBu)MnVN to ammonia and a Mn(II) complex has been accomplished using 9,10-dihydroacridine and a combination of an appropriately matched photoredox catalyst and weak Brønsted acid. Acid-reductant pairs with effective bond dissociation free energies between 35 and 46 kcal/mol exhibited high efficiencies. This light-driven method may provide a blueprint for new approaches to catalytic homogeneous ammonia synthesis under ambient conditions. |  |
182 | Oxidative Addition of Dihydrogen, Boron Compounds, and Aryl Halides to a Cobalt(I) Cation Supported by a Strong-Field Pincer LigandStephan M. Rummelt, Hongyu Zhong, Nadia G. Léonard, Scott P. Semproni and Paul J. Chirik Organometallics 2019, 38, 1081–1090. AbstractCationic cobalt(I) dinitrogen complexes with a strong-field tridentate pincer ligand were prepared, and the oxidative addition of polar and nonpolar bonds was studied. Addition of H2 to [(iPrPNP)Co(N2)]+ (iPrPNP = 2,6-bis((diisopropylphosphaneyl)methyl)pyridine) in deuterated tetrahydrofuran (THF) resulted in rapid oxidative addition and formation of the cis-Co(III) dihydride complex, cis-[(iPrPNP)Co(H)2L]+, where L = THF or N2. The addition of H2 was reversible as evidenced by the dynamics observed by variable-temperature 1H NMR spectroscopy and the regeneration of [(iPrPNP)Co(N2)]+ upon exposure to dinitrogen. In contrast, addition of HBPin (Pin = pinacolato), B2Pin2, and aryl halides resulted in the formation of net one-electron oxidation products: cationic Co(II)–boryl and Co(II)–halide/aryl complexes, respectively. All products were structurally characterized by X-ray crystallography, and the electronic structures were determined by a combination of magnetic moment measurements, electron paramagnetic resonance spectroscopy, and density functional theory calculations. Monitoring the addition of HBPin to [(iPrPNP)Co(N2)]+ provided evidence for a transient Co(III) oxidative addition product that likely undergoes comproportionation with the cobalt(I) starting material to generate the observed Co(II) products. | 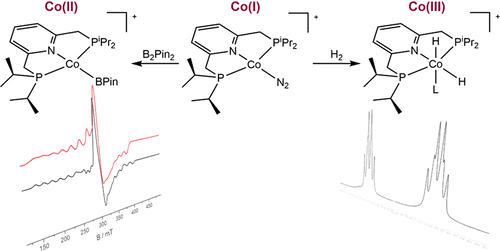 |
181 | Exploring C(sp3)–C(sp3) reductive elimination from an isolable iron metallacycleMatthew V. Joannou, Jonathan M. Darmon, Máté J. Bezdek, and Paul J. Chirik Polyhedron 2019, 159, 308–317. AbstractA six-coordinate iron metallacyclopentane, (phen)2Fe(CH2)4, supported by two 1,10-phenanthroline (phen) ligands, has been synthesized and structurally and spectroscopically characterized. The complex is diamagnetic and an idealized octahedral geometry was observed in the solid state. The electronic structure of (phen)2Fe(CH2)4 was determined by a combination of X-ray diffraction, Mössbauer spectroscopy, and DFT analyses and is best described as a low-spin Fe(III) center antiferromagnetically coupled to a radical anion delocalized equally over both phen ligands. The reactivity of (phen)2Fe(CH2)4 under different conditions was explored. Thermolysis or photolysis promoted elimination reactions and mixtures of isomeric butenes and butane were observed. Reactions of (phen)2Fe(CH2)4 with ethylene and isoprene yielded 3% and 11% of reductive elimination product cyclobutane, respectively, along with butane and butene isomers. Addition of π-accepting ligands such as carbon monoxide, maleic anhydride, or 1,4-benzoquinone to (phen)2Fe(CH2)4 promoted C(sp3)-C(sp3) reductive elimination as judged by high selectivity for cyclobutane formation. Two electron oxidation of (phen)2Fe(CH2)4 with two equivalents of ferrocenium tetraphenylborate also exclusively yielded cyclobutane in 95% yield. The electronic structure and reactivity of related bis(bipyridine) iron dialkyl compounds previously isolated by Kochi and co-workers were also revisited and their electronic structures revised based on structural, spectroscopic and computational data. | 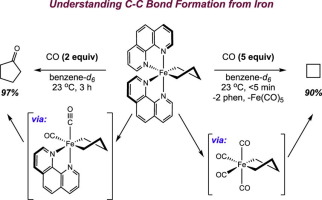 |
180 | Dinitrogen Coupling to a Terpyridine-Molybdenum Chromophore Is Switched on by Fermi ResonanceShahnawaz Rafiq, Máté J. Bezdek, Paul J. Chirik and Gregory D. Scholes AbstractThe traditional view of a chemical change is inherently local and classical, and such a change relies on a mix of thermodynamic and kinetic parameters to control reactivity. Often, the thermodynamic stability of chemical bonds necessitates significant energy input for activation. One fundamental question is potentially transformative: can quantum mechanics enable selective bond activation? A possible approach involves strategic input of energy to reaction-specific vibrational levels. Toward this goal, our work describes the coupling of vibrational motions in a terpyridine-molybdenum complex hosting a nonreactive substrate—dinitrogen. Ultrafast coherence spectroscopies revealed a Fermi-resonance coupling mechanism connecting in-plane breathing motion of the light-harvesting terpyridines with the stretching motion of the spatially disparate dinitrogen bridge. Notably, the coupling is significantly enhanced in the photoexcited state. This Fermi resonance indicates an energy conduit that drives the two motions in sync and thereby amplifies vibrational energy exchange. Achieving selective bond activation by bridging vibrations could present a quantum-inspired design principle in synthetic chemistry. |  |
179 | Exploring the Alcohol Stability of Bis(phosphine) Cobalt Dialkyl Precatalysts in Asymmetric Alkene HydrogenationHongyu Zhong, Max R. Friedfeld, Jeffrey Camacho-Bunquin, Hyuntae Sohn, Ce Yang, Massimiliano Delferro, and Paul J. Chirik Organometallics 2018, 38, 149–156. AbstractCobalt complexes bearing enantiopure, bidentate bis(phosphine) ligands exhibit extraordinary activity and stereoselectivity for the hydrogenation of enamides. Optimal performance was observed in polar protic solvents such as methanol, an industrially preferred green solvent but a medium that is often a poison for reduced Earth abundant metals. The interaction of the low-spin cobalt(II) dialkyl complex, (R,R)-(iPr-DuPhos)Co(CH2SiMe3)2, with alcohols including 4-methoxyphenol, pinacol, and CH3OH was studied. With the alcohols lacking β-hydrogens, cobalt bis(alkoxide) complexes were isolated and structurally characterized. With methanol, protonolysis of the alkyl ligands was again observed followed by dehydrogenation of the alcohol and [(R,R)-(iPr-DuPhos)Co]2(μ-CO)2 was isolated. Both solid-state and solution EXAFS studies were conducted to establish the spectroscopic signatures of bis(phosphine) cobalt(II) and cobalt(0) complexes relevant to catalytic hydrogenation and also to probe the role of phosphine dissociation in methanol. |  |
178 | Cobalt Pincer Complexes in Catalytic C–H Borylation: The Pincer Ligand Flips Rather Than DearomatizesHaixia Li, Jennifer V. Obligacion, Paul J. Chirik, and Michael B. Hall ACS Catal. 2018, 8, 10606–10618. AbstractThe mechanism for the borylation of an aromatic substrate by a cobalt pincer complex was investigated by density functional theory calculations. Experimental observations identified trans-(iPrPNP)CoH2(BPin) as the resting state in the borylation of five-membered heteroarenes and 4-BPin-(iPrPNP)Co(N2)BPin as the resting state in the catalytic borylation of arene substrates. The active species, 4-R-(iPrPNP)CoBPin (R = H, BPin), were generated by reductive elimination of H2 in the former, through Berry pseudorotation to the cis isomer and N2 loss in the latter. The catalytic mechanism of the resulting Co(I) complex was computed to involve three main steps: C–H oxidative addition of the aromatic substrate (C6H6), reductive elimination of PhBPin, and regeneration of the active complex. The oxidative addition product formed through the most favorable pathway, where the breaking C–H bond of C6H6 is parallel to a line between the two phosphine atoms, leaves the complex with a distorted PNP ligand, which rearranges to a more stable complex via dissociation and reassociation of HBPin. Alternative pathways, σ-bond metathesis, and the oxidative addition in which the breaking C–H bond is parallel to the Co–B bond are predicted to be unlikely for this Co(I) complex. The thermodynamically favorable formation of the product PhBPin via reductive elimination drives the reaction forward. The active species regenerates through the oxidative addition of B2Pin2 and reductive elimination of HBPin. In the overall reaction, the flipping (refolding) of the five-membered phosphine rings, which connects the species with two phosphine rings folded in the same direction and that with them folded in different directions, is found to play an important role in the catalytic process, as it relieves steric crowding within the PNP ligand and opens Co coordination space. Metal–ligand cooperation based on the ligand’s aromatization/dearomatization, a common mechanism for heavy-metal pincer complexes, and the dissociation of one phosphine ligand do not apply in this system. This study provides guidance for understanding important features of pincer ligands with first-transition-row metals that differ from those in heavier metal complexes. |  |
177 | Proton-Coupled Electron Transfer to a Molybdenum Ethylene Complex Yields a β-Agostic Ethyl: Structure, Dynamics and MechanismMáté J. Bezdek and Paul J. Chirik J. Am. Chem. Soc. 2018, 140, 13817–13826. AbstractThe interconversion of molybdenum ethylene and ethyl complexes by proton-coupled electron transfer (PCET) is described, an unusual transformation in organometallic chemistry. The cationic molybdenum ethylene complex [(PhTpy)(PPh2Me)2Mo(C2H4)][BArF24] ([1-C2H4]+; PhTpy = 4′-Ph-2,2′,6′,2″-terpyridine, ArF24 = [C6H3-3,5-(CF3)2]4) was synthesized, structurally characterized, and its electronic structure established by a combination of spectroscopic and computational methods. The overall electronic structure is best described as a molybdenum(III) complex with a metallacyclopropane and a redox neutral terpyridine ligand. Addition of the nonclassical ammine complex [(PhTpy)(PPh2Me)2Mo(NH3)][BArF24] ([1-NH3]+) to [1-C2H4]+ resulted in a net C–H bond-forming PCET reaction to yield the molybdenum ethyl [(PhTpy)(PPh2Me)2Mo(CH2CH3)][BArF24] ([1-CH2CH3]+) and amido [(PhTpy)(PPh2Me)2Mo(NH2)][BArF24] ([1-NH2]+) compounds. The reaction was reversed by addition of 2,4,6-tritert-butylphenoxyl radical to [1-CH2CH3]+. The solid-state structure of [1-CH2CH3]+ established a β-agostic ethyl ligand that is maintained in solution as judged by variable temperature 1H and 13C NMR experiments. A combination of variable-temperature NMR experiments and isotopic labeling studies were used to probe the dynamics of [1-CH2CH3]+ and established restricted β-agostic −CH3 rotation at low temperature (ΔG‡ = 9.8 kcal mol–1 at −86 °C) as well as ethyl isomerization by β-hydride elimination-olefin rotation-reinsertion (ΔH‡ = 19.3 ± 0.6 kcal mol–1; ΔS‡ = 3.4 ± 1.7 cal mol–1 K–1). The β-(C–H) bond-dissociation free energy (BDFE) in [1-CH2CH3]+ was determined experimentally as 57 kcal mol–1 (THF) supported by a DFT-computed value of 52 kcal/mol–1 (gas phase). Comparison of pKa and electrochemical data for the complexes [1-C2H4]+ and [1-NH3]+ in combination with a deuterium kinetic isotope effect (kH/kD) of 3.5(2) at 23 °C support a PCET process involving initial electron transfer followed by protonation leading to the formation of [1-CH2CH3]+ and [1-NH2]+ or a concerted pathway. The data presented herein provides a structural, thermochemical and mechanistic foundation for understanding the PCET reactivity of organometallic complexes with alkene and alkyl ligands. | 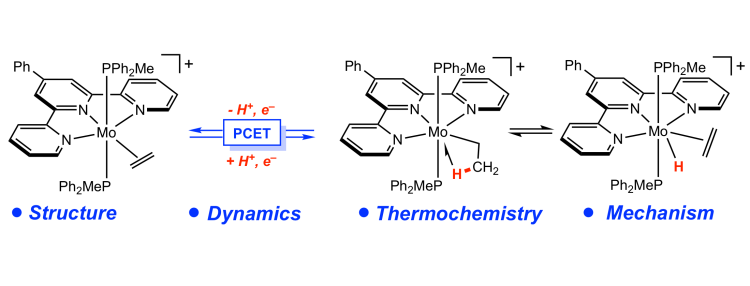 |
176 | Site-Selective Nickel-Catalyzed Hydrogen Isotope Exchange in N-Heterocycles and Its Application to the Tritiation of PharmaceuticalsHaifeng Yang, Cayetana Zarate, W. Neil Palmer, Nelo Rivera, David Hesk, and Paul J. Chirik ACS Catal. 2018, 8, 10210–10218. AbstractA nickel-catalyzed method for the site-selective hydrogen isotope exchange (HIE) of C(sp2)–H bonds in nitrogen heteroarenes is described and applied to the tritiation of pharmaceuticals. The α-diimine nickel hydride complex [(iPrDI)Ni(μ2–H)]2 (iPrDI = N,N′-bis(2,6-diisopropylphenyl)-2,3-butanediimine) mediates efficient HIE when employed as a single component precatalyst or generated in situ from readily available and air-stable metal and ligand precursors (iPrDI, [(NEt3)Ni(OPiv)2]2 (Piv = pivaloyl) and (EtO)3SiH). The nickel catalyst offers distinct advantages over existing methods, including: (i) high HIE activity at low D2 or T2 pressure; (ii) tolerance of functional groups, including aryl chlorides, alcohols, secondary amides, and sulfones; (iii) activity with nitrogen-rich molecules such as the chemotherapeutic imatinib; and (iv) the ability to promote HIE in sterically hindered positions generally inaccessible with other transition metal catalysts. Representative active pharmaceutical ingredients were tritiated with specific activities in excess of the thresholds required for drug absorption, distribution, metabolism, and excretion studies (1 Ci/mmol) and for protein receptor–ligand binding assays (15 Ci/mmol). The activity and selectivity of the nickel-catalyzed method are demonstrated by comparison with the current state-of-the-art single-site (iridium and iron) and heterogeneous (Raney nickel and rhodium black) catalysts. A pathway involving C(sp2)–H activation by a α-diimine nickel hydride monomer is consistent with the experimentally measured relative rate constants for HIE with electronically disparate pyridines, the pressure-dependence of activity, positional selectivity preferences, and kinetic isotope effects. |  |
175 | Iron-Mediated Coupling of Carbon Dioxide and Ethylene: Macrocyclic Metallalactones Enable Access to Various CarboxylatesStephan M. Rummelt, Hongyu Zhong, Ilia Korobkov, and Paul J. Chirik J. Am. Chem. Soc. 2018, 140, 11589–11593. AbstractTreatment of (iPrPDI)Fe(N2)2 (iPrPDI, 2,6-(2,6-iPr2C6H3N═CMe)2C5H3N) with CO2 and ethylene resulted in the formation of a homologous series of saturated and unsaturated iron carboxylate products, (iPrPDI)Fe(O2CR), the distribution of which depends on the ratio of the reagents. The solid-state and electronic structures of a saturated product, (iPrPDI)Fe(O2CC2H5), were elucidated. Product distributions, deuterium labeling studies, and stoichiometric experiments support initial formation of a five-membered metallalactone intermediate, which undergoes subsequent ethylene insertions to generate macrocyclic metallalactones. Competitive β-hydride elimination, CO2 insertion, or reaction with H2 determines the fate of the metallalactone, the latter accounting for formation of iron complexes with saturated carboxylates. Similar reactivity was observed upon addition of propiolactone and ethylene to (iPrPDI)Fe(N2)2, supporting C–O oxidative addition and C–C bond formation through metallacycle intermediates. | 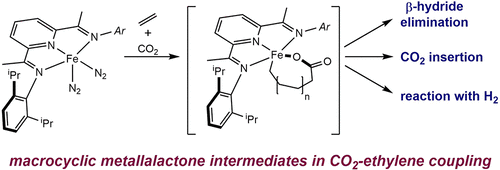 |
174 | Synthesis and Reactivity of Reduced α-Diimine Nickel Complexes Relevant to Acrylic Acid SynthesisMatthew V. Joannou, Máté J. Bezdek, Khalid Albahily, Ilia Korobkov, and Paul J. Chirik Organometallics 2018, 37, 3389–3393. AbstractThe aryl-substituted α-diimine (DI) nickel vinyl complex (iPrDI)Ni(CH═CH2) (iPrDI = [2,6-(iPr)2C6H3N═C(CH3)]2) was synthesized and structurally characterized. The complex is dimeric in the solid state and has a distorted-square-planar geometry at nickel. A combination of single-crystal X-ray diffraction, EPR, magnetic susceptibility, and NMR analyses was used to elucidate the electronic structure of the compound, and it is best described as a low-spin Ni(II) derivative with a singly reduced α-diimine chelate. Addition of CO2 to the nickel vinyl complex resulted in insertion into the nickel–carbon bond to yield the corresponding nickel acrylate (iPrDI)Ni(κ2-O2CCH═CH2). EPR spectroscopy coupled with DFT calculations established that the S = 1/2 product maintains the nickel(II) oxidation state with an α-diimine-centered radical. Addition of acrylic acid to (iPrDI)Ni(CH═CH2) induced rapid, net bimetallic reductive elimination to release butadiene and produced the metastable olefin-bound acrylic acid complex (iPrDI)Ni(η2-CH2═CHCO2H). Over the course of 2 h at 23 °C, this complex underwent a net oxidation to produce (iPrDI)Ni(κ2-O2CCH═CH2), with concomitant loss of H2. | 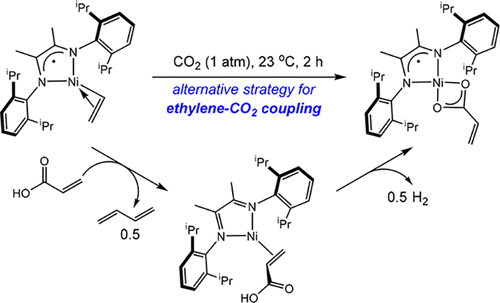 |
173 | Cobalt-catalyzed asymmetric hydrogenation of enamides enabled by single-electron reductionMax R. Friedfeld, Hongyu Zhong, Rebecca T. Ruck, Michael Shevlin and Paul J. Chirik AbstractIdentifying catalyst activation modes that exploit one-electron chemistry and overcome associated deactivation pathways will be transformative for developing first-row transition metal catalysts with performance equal or, ideally, superior to precious metals. Here we describe a zinc-activation method compatible with high-throughput reaction discovery that identified scores of cobalt-phosphine combinations for the asymmetric hydrogenation of functionalized alkenes. An optimized catalyst prepared from (R,R)-Ph-BPE {Ph-BPE, 1,2-bis[(2R,5R)-2,5-diphenylphospholano]ethane} and cobalt chloride [CoCl2·6H2O] exhibited high activity and enantioselectivity in protic media and enabled the asymmetric synthesis of the epilepsy medication levetiracetam at 200-gram scale with 0.08 mole % catalyst loading. Stoichiometric studies established that the cobalt (II) catalyst precursor (R,R)-Ph-BPECoCl2 underwent ligand displacement by methanol, and zinc promoted facile one-electron reduction to cobalt (I), which more stably bound the phosphine. | |
172 | Cobalt-catalysed alkene hydrogenation: a metallacycle can explain the hydroxyl activating effect and the diastereoselectivityGlenn R. Morello, Hongyu Zhong, Paul J. Chirik and Kathrin H. Hopmann AbstractBis(phosphine)cobalt dialkyl complexes have been reported to be highly active in the hydrogenation of tri-substituted alkenes bearing hydroxyl substituents. Alkene substrates containing ether, ester, or ketone substituents show minimal reactivity, indicating an activating effect of the hydroxyl group. The mechanistic details of bis(phosphine)cobalt-catalysed hydrogenation were recently evaluated computationally (X. Ma, M. Lei, J. Org. Chem. 2017, 82, 2703–2712) and a Co(0)–Co(II) redox mechanism was proposed. However, the activating effect of the hydroxyl substituent and the accompanying high diastereoselectivity were not studied. Here we report a computational study rationalizing the role of the hydroxyl group through a key metallacycle species. The metallacycle is part of a non-redox catalytic pathway proceeding through Co(II) intermediates throughout. The preference for alcohol over ether substrates and the high diastereoselectivity of terpinen-4-ol hydrogenation are correctly predicted in computations adopting the new pathway, whereas the alternative redox mechanism predicts ethers rather than alcohols to be more reactive substrates. Additional experimental evidence supports the role of the hydroxyl group in the metallacycle mechanism. Our work highlights the importance of employing known substrate preferences and stereoselectivities to test the validity of computationally proposed reaction pathways. |  |
171 | Ultrafast Photophysics of a Dinitrogen-Bridged Molybdenum ComplexShahnawaz Rafiq , Máté J. Bezdek, Marius Koch , Paul J. Chirik, and Gregory D. Scholes J. Am. Chem. Soc. 2018, 140, 6298–6307. AbstractAmong the many metal–dinitrogen complexes synthesized, the end-on bridging (μ2, η1, η1—N2) coordination mode is notoriously unreactive for nitrogen fixation. This is principally due to the large activation energy for ground-state nitrogen–element bond formation and motivates exploration of the photoexcited reactivity of this coordination mode. To provide the foundation for this concept, the photophysics of a dinitrogen-bridged molybdenum complex was explored by ultrafast electronic spectroscopies. The complex absorbs light from the UV to near-IR, and the transitions are predominantly of metal-to-ligand charge transfer (MLCT) character. Five excitation wavelengths (440, 520, 610, 730, and 1150 nm) were employed to access MLCT bands, and the dynamics were probed between 430 and 1600 nm. Despite the large energy space occupied by electronic states (ca. 1.2 eV), the dynamics were independent of the excitation wavelength. In the proposed kinetic model, photoexcitation from a Mo–N═N–Mo centered ground state populates the π*-state delocalized over two terpyridine ligands. Due to a large terpyridine–terpyridine spatial separation, electronic localization occurs within 100 fs, augmented by symmetry breaking. The subsequent interplay of internal conversion and intersystem crossing (ISC) populates the lowest 3MLCT state in 2–3 ps. Decay to the ground state occurs either directly or via a thermally activated metal-centered (3MC) trap state having two time constants (10–15 ps, 23–26 ps [298 K]; 103 ps, 612 ps [77 K]). ISC between 1MLCT and 3MLCT involves migration of energized electron density from the terpyridine π* orbitals to the Mo–N═N–Mo core. Implication of the observed dynamics for the potential N–H bond forming reactivity are discussed. |  |
170 | Pyridine(diimine) Molybdenum-Catalyzed Hydrogenation of Arenes and Hindered Olefins: Insights into Precatalyst Activation and Deactivation PathwaysMatthew V. Joannou, Máté J. Bezdek, and Paul J. Chirik AbstractPyridine(diimine) molybdenum bis(olefin) and bis(alkyl) complexes were synthesized, characterized, and examined for their catalytic activity in the hydrogenation of benzene and a selection of substituted arenes. The molybdenum bis(alkyl) complex (4-tBu-iPrPDI)Mo(CH2SiMe3)2 (iPrPDI = 2,6-(2,6-(C(CH3)2H)2C6H3N═CMe)2C5H3N) exhibited the highest activity for the hydrogenation of benzene, producing cyclohexane in >98% yield at 23 °C under 4 atm of hydrogen after 48 h. Toluene and o-xylene were similarly hydrogenated to their respective cycloalkanes, with the latter yielding predominantly (79:21 dr) cis-1,2-dimethylcyclohexane. The molybdenum-catalyzed hydrogenation of naphthalene yielded tetralin exclusively, and this selectivity was maintained at higher H2 pressure. At 32 atm of H2, more hindered arenes such as monosubstituted benzenes, biphenyl, and m– and p-xylenes underwent hydrogenation with yields ranging between 20 and >98%. (4-tBu-iPrPDI)Mo(CH2SiMe3)2 was also a competent alkene hydrogenation catalyst, supporting stepwise reduction of benzene to cyclohexadiene and cyclohexene during molybdenum-catalyzed arene hydrogenation. Deuterium labeling studies for the molybdenum-catalyzed hydrogenation of benzene produced numerous isotopologues and stereoisomers of cyclohexane, indicating reversible hydride (deuteride) insertion/β-H(D) elimination, diene/olefin binding, and allylic C–H(D) activation during the reaction. The resting state of the catalyst under neat conditions was established as the η6-benzene complex (iPrPDI)Mo(η6-benzene). Under catalytic conditions, pyridine underwent C–H activation of the 2-position and furan underwent formal C–O oxidative addition to yield a “metallapyran”. Both reactions were identified as important catalyst deactivation pathways for the attempted molybdenum-catalyzed hydrogenation of heteroarenes. |  |
169 | Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroborationJennifer V. Obligacion and Paul J. Chirik Nat. Rev. Chem. 2018, 2, 15–34. AbstractThe addition of X3Si–H or X2B–H (X = H, OR or R) across a C–C multiple bond is a well-established method for incorporating silane or borane groups, respectively, into hydrocarbon feedstocks. These hydrofunctionalization reactions are often mediated by transition metal catalysts, with precious metals being the most commonly used owing to the ability to optimize reaction scope, rates and selectivities. For example, platinum catalysts effect the hydrosilylation of alkenes with anti-Markovnikov selectivity and constitute an enabling technology in the multibillion dollar silicones industry. Increased emphasis on sustainable catalytic methods and on more economic processes has shifted the focus to catalysis with more earth-abundant transition metals, such as iron, cobalt and nickel. This Review describes the use of first-row transition metal complexes in catalytic alkene hydrosilylation and hydroboration. Defining advances in the field are covered, noting the chemistry that is unique to first-row transition metals and the design features that enable them to exhibit precious-metal-like reactivity. Other important features, such as catalyst activity and stability, are covered, as are practical considerations, such as cost and safety. | 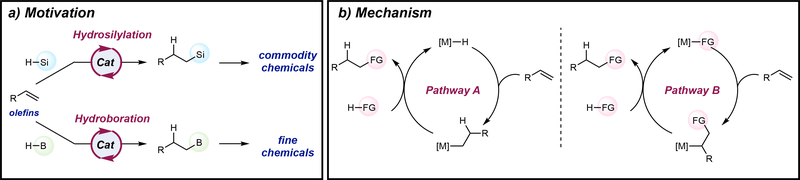 |
168 | Synthesis and Electronic Structure Diversity of Pyridine(diimine)iron Tetrazene ComplexesAmanda C. Bowman, Aaron M. Tondreau, Emil Lobkovsky, Grant Margulieux, and Paul J. Chirik Inorg. Chem. 2018, 57, 9634–9643. AbstractA series of pyridine(diimine)iron tetrazene compounds, (iPrPDI)Fe[(NR)NN(NR)] [iPrPDI = 2,6-(ArN = CMe)2C5H3N; Ar = 2,6-iPr2C6H3] has been prepared either by the addition of 2 equiv of an organic azide, RN3, to the corresponding iron bis(dinitrogen) compound, (iPrPDI)Fe(N2)2 or by the addition of azide to the iron imide derivatives, (iPrPDI)FeNR. The electronic structures of these compounds were determined using a combination of metrical parameters from X-ray diffraction, solution and solid-state magnetic measurements, zero-field 57Fe Mössbauer and 1H NMR spectroscopies, and density functional theory calculations. The overall electronic structure of the iron tetrazene compounds is sensitive to the nature of the tetrazene nitrogen substituent with three distinct classes of compounds identified: (i) overall diamagnetic (S = 0) compounds arising from intermediate-spin iron(II) centers (SFe = 1) engaged in antiferromagnetic coupling with both pyridine(diimine) and tetrazene radical anions (SPDI = −1/2 and Stetrazene = −1/2; R = 2-adamantyl, cyclooctyl, benzyl); (ii) overall S = 1 compounds best described as intermediate-spin iron(III) (SFe = 3/2) derivatives engaged in antiferromagnetic coupling with a pyridine(diimine) radical anion (SPDI = −1/2; R = 3,5-Me2C6H3, 4-MeC6H4); (iii) overall S = 2 compounds best described as high-spin iron(III) centers (SFe = 5/2) engaged in antiferromagnetic coupling to a pyridine(diimine) radical anion (SPDI = −1/2; R = 1-adamantyl). For both the intermediate- and high-spin ferric cases, the tetrazene ligand adopts the closed-shell, dianionic form, [N4R2]2–. For the case where R = SiMe3, spin-crossover behavior is observed, arising from a spin-state change from intermediate- to high-spin iron(III). |  |
167 | Selective [1,4]-Hydrovinylation of 1,3-Dienes with Unactivated Olefins Enabled by Iron Diimine CatalystsValerie A. Schmidt, C. Rose Kennedy, Máté J. Bezdek, and Paul J. Chirik J. Am. Chem. Soc. 2018, 140, 3443–3453. AbstractThe selective, intermolecular [1,4]-hydrovinylation of conjugated dienes with unactivated α-olefins catalyzed by α-diimine iron complexes is described. Value-added “skipped” diene products were obtained with exclusive [1,4]-selectivity, and the formation of branched, (Z)-olefin products was observed with no evidence for alkene isomerization. Mechanistic studies conducted with the well-defined, single-component iron precatalyst (MesDI)Fe(COD) (MesDI = [2,4,6-Me3-C6H2-N═CMe]2); COD = 1,5-cyclooctadiene) provided insights into the origin of the high selectivity. An iron diene complex was identified as the catalyst resting state, and one such isoprene complex, (iPrDI)Fe(η4-C5H8), was isolated and characterized. A combination of single crystal X-ray diffraction, Mößbauer spectroscopy, magnetic measurements, and DFT calculations established that the complex is best described as a high-spin Fe(I) center (SFe = 3/2) engaged in antiferromagnetic coupling to an α-diimine radical anion (SDI = −1/2), giving rise to the observed S = 1 ground state. Deuterium-labeling experiments and kinetic analyses of the catalytic reaction provided support for a pathway involving oxidative cyclization of an alkene with the diene complex to generate an iron metallacycle. The observed selectivity can be understood in terms of competing steric interactions in the transition states for oxidative cyclization and subsequent β-hydrogen elimination. | ![Selective [1,4]-Hydrovinylation of 1,3-Dienes with Unactivated Olefins Enabled by Iron Diimine Catalysts](/wp-content/uploads/2022/10/img-publ-167.png) |
166 | Interconversion of Molybdenum Imido and Amido Complexes by Proton-Coupled Electron TransferMáté J. Bezdek and Paul J. Chirik Angew. Chem. Int. Ed. 2018, 57, 2224–2228. AbstractInterconversion of the molybdenum amido [(PhTpy)(PPh2Me)2Mo(NHtBuAr)][BArF24] (PhTpy=4′-Ph-2,2′,6′,2“-terpyridine; tBuAr=4-tert-butyl-C6H4; ArF24=(C6H3-3,5-(CF3)2)4) and imido [(PhTpy)(PPh2Me)2Mo(NtBuAr)][BArF24] complexes has been accomplished by proton-coupled electron transfer. The 2,4,6-tri-tert-butylphenoxyl radical was used as an oxidant and the non-classical ammine complex [(PhTpy)(PPh2Me)2Mo(NH3)][BArF24] as the reductant. The N−H bond dissociation free energy (BDFE) of the amido N−H bond formed and cleaved in the sequence was experimentally bracketed between 45.8 and 52.3 kcal mol−1, in agreement with a DFT-computed value of 48 kcal mol−1. The N−H BDFE in combination with electrochemical data eliminate proton transfer as the first step in the N−H bond-forming sequence and favor initial electron transfer or concerted pathways. |  |
165 | Air-Stable α-Diimine Nickel Precatalysts for the Hydrogenation of Hindered, Unactivated AlkenesNadia G. Léonard and Paul J. Chirik AbstractTreatment of a mixture of air-stable nickel(II) bis(octanoate), Ni(O2CC7H15)2, and α-diimine ligand, iPrDI or CyADI (iPrDI = [2,6-iPr2–C6H3N═C(CH3)]2, CyADI = [C6H11N═C(CH3)]2) with pinacolborane (HBPin) generated a highly active catalyst for the hydrogenation of hindered, essentially unfunctionalized alkenes. A range of tri- and tetrasubstituted alkenes was hydrogenated and a benchtop procedure for the hydrogenation of 1-phenyl-1-cyclohexene on a multigram scale was demonstrated and represents an advance in catalyst activity and scope for the nickel-catalyzed hydrogenation of this challenging class of alkenes. Deuteration of 1,2-dimethylindene with the in situ-generated nickel catalyst with iPrDI exclusively furnished the 1,2-syn–d2-dimethylindane. With cyclic trisubstituted alkenes, such as 1-methyl-indene and methylcyclohexene, deuteration with the in situ generated nickel catalyst under 4 atm of D2 produced multiple deuterated isotopologues of the alkanes, signaling chain running processes that are competitive with productive hydrogenation. Stoichiometric studies, titration, and deuterium labeling experiments identified that the borane reagent served the dual role of reducing nickel(II) bis(carboxylate) to the previously reported nickel hydride dimer [(iPrDI)NiH]2 and increasing the observed hydrogenation activity. Performing the catalyst activation procedure with D2 gas and HBPin generated both HD and DBPin, establishing that the borane is involved in H2 activation as judged by 1H and 11B nuclear magnetic resonance spectroscopies. |  |
164 | Synthesis and Reactivity of Pyridine(diimine) Molybdenum Olefin Complexes: Ethylene Dimerization and Alkene DehydrogenationMatthew V. Joannou, Máté J. Bezdek, Khalid Al-Bahily, Ilia Korobkov, and Paul J. Chirik Organometallics 2017, 36, 4215–4223. AbstractReduced pyridine(diimine) molybdenum olefin complexes have been synthesized and structurally characterized. Examples with 1,5-cyclooctadiene, (PDI)Mo(η2:η2-1,5-COD) (COD = 1,5-cyclooctadiene) adopt a distorted-trigonal-bipyramidal geometry and are best described as low-spin Mo(II) compounds arising from significant π back-donation to the ligand from a reduced molybdenum center. With the 2,6-diisopropyl N-aryl-substituted variant of the pyridine(diimine) ligand, a molybdenum bis(ethylene) complex was obtained. Reducing the size of the N-aryl substituents to 2,4,6-trimethyl resulted in isolation of (MesPDI)Mo(η4-butadiene)(η2-ethylene) following sodium amalgam reduction of the corresponding molybdenum(III) trichloride complex in the presence of excess ethylene. Analysis of the byproducts of the reaction and olefin addition experiments demonstrate that butadiene formation is consistent with a pathway involving ethylene coupling to form 1-butene followed by allylic dehydrogenation to produce butadiene. Excess ethylene serves as the hydrogen acceptor. The dehydrogenation reaction was also compatible with α-olefins, as reduction of either (iPrPDI)MoCl3 or (MesPDI)MoCl3 in the presence of 1-hexene resulted in isolation of (PDI)Mo(η4-1,3-hexadiene)(η2-1-hexene) complexes. An α,ω-diene complex, (iPrPDI)Mo(η2:η2-1,6-heptadiene), was also synthesized and importantly displayed no cycloaddition chemistry, suggesting that first-row metal pyridine(diimine) complexes are thus far unique in promoting cyclobutane synthesis. | 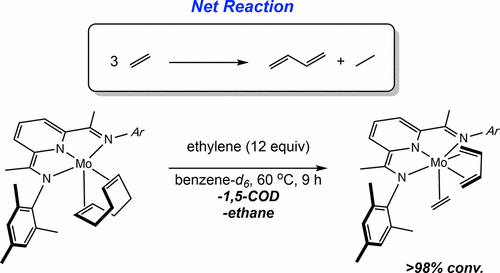 |
163 | Insights into Activation of Cobalt Pre-Catalysts for C(sp2)–H FunctionalizationJennifer V. Obligacion, Hongyu Zhong, and Paul J. Chirik Israel Journal of Chemistry 2017, 57, 1032–1036. AbstractThe activation of readily prepared, air-stable cobalt(II) bis(carboxylate) pre-catalysts for the functionalization of C(sp2)−H bonds has been systematically studied. With the pyridine bis(phosphine) chelate, iPrPNP, treatment of 1–(O2CtBu)2 with either B2Pin2 or HBPin generated cobalt boryl products. With the former, reduction to (iPrPNP)CoIBPin was observed while with the latter, oxidation to the cobalt(III) dihydride boryl, trans-(iPrPNP)Co(H)2BPin occurred. The catalytically inactive cobalt complex, Co[PinB(O2CtBu)2]2, accompanied formation of the cobalt-boryl products in both cases. These results demonstrate that the pre-catalyst activation from cobalt(II) bis(carboxylates), although effective and utilizes an air-stable precursor, is less efficient than activation of cobalt(I) alkyl or cobalt(III) dihydride boryl complexes, which are quantitatively converted to the catalytically relevant cobalt(I) boryl. Related cobalt(III) dihydride silyl and cobalt(I) silyl complexes were also synthesized from treatment of trans-(iPrPNP)Co(H)2BPin and (iPrPNP)CoPh with HSi(OEt)3, respectively. No catalytic silylation of arenes was observed with either complex likely due to the kinetic preference for reversible C−H reductive elimination rather than product- forming C−Si bond formation from cobalt(III). Syntheses of the cobalt(II) bis(carboxylate) and cobalt(I) alkyl of iPrPONOP, a pincer where the methylene spacers have been replaced by oxygen atoms, were unsuccessful due to deleterious P−O bond cleavage of the pincer. Despite their structural similarity, the rich catalytic chemistry of iPrPNP was not translated to iPrPONOP due to the inability to access stable cobalt precursors as a result of ligand decomposition via P−O bond cleavage. |  |
162 | Cobalt-Catalyzed Stereoretentive Hydrogen Isotope Exchange of C(sp3)–H BondsW. Neil Palmer and Paul J. Chirik ACS Catal. 2017, 7, 5674–5678. AbstractCobalt dialkyl complexes bearing α-diimine ligands proved to be active precatalysts for the nondirected, C(sp3)–H selective hydrogen isotope exchange (HIE) of alkylarenes using D2 gas as the deuterium source. Alkylarenes with a variety of substitution patterns and heteroatom substituents on the arene ring were successfully labeled, enabling high levels of incorporation into primary, secondary, and tertiary benzylic C(sp3)–H bonds. In some cases, the HIE proceeded with high diastereoselectivity and application of the cobalt-catalyzed method to enantioenriched substrates with benzylic stereocenters provided enantioretentive hydrogen isotope exchange at tertiary carbons. |  |
161 | Synthesis of Iron Hydride Complexes Relevant to Hydrogen Isotope Exchange in PharmaceuticalsRenyuan Pony Yu, Jonathan M. Darmon, Scott P. Semproni, Zoë R. Turner, and Paul J. Chirik Organometallics 2017, 36, 4341–4343. AbstractAddition of H2 gas to the bis(arylimidazolin-2-ylidene)pyridine iron bis(dinitrogen) complex (H4–iPrCNC)Fe(N2)2 resulted in facile oxidative addition of the H–H bond to yield a mixture of (H4–iPrCNC)FeH4 and trans-(H4–iPrCNC)FeH2(N2), depending on the partial pressures of H2 and N2. Both iron hydride complexes were characterized by X-ray diffraction and proved relevant to the catalytic hydrogen isotope exchange of arene C(sp2)–H bonds. Activation of the benzene-d6 solvent at ambient temperature produced deuterated isotologues of both compounds that exhibit large isotopic perturbation of resonances in the hydride signals. |  |
160 | Determining and Understanding N-H Bond Strengths in Synthetic Nitrogen Fixation CyclesMáté J. Bezdek, Iraklis Pappas, and Paul J. Chirik Top. Organomet. Chem. 2017, 60, 1–21. AbstractThe fixation of atmospheric dinitrogen to ammonia using molecular catalysts has been a long-standing challenge in homogeneous catalysis and synthetic chemistry. New approaches to this problem may offer more energy-efficient and carbon-neutral routes to this important industrial compound. Despite the ubiquity of ammine, amide, imide and diazenide ligands in coordination chemistry, little thermodynamic data is available for understanding N-H bond strengths in molecules bearing these nitrogenous fragments. This article presents an overview of both computational and experimental approaches for the determination of N-H bond dissociation free energies in a variety of compounds relevant to nitrogen fixation to ammonia. The influence of metal oxidation state, ancillary ligand and identity of the nitrogen donor are highlighted. Implications for future design of molecular systems for the reduction of dinitrogen are discussed. | |
159 | Mechanistic Studies of Cobalt-Catalyzed C(sp2)–H Borylation of Five-Membered Heteroarenes with PinacolboraneJennifer V. Obligacion and Paul J. Chirik ACS Catal. 2017, 7, 4366–4371. AbstractStudies into the mechanism of cobalt-catalyzed C(sp2)–H borylation of five-membered heteroarenes with pinacolborane (HBPin) as the boron source established the catalyst resting state as the trans-cobalt(III) dihydride boryl, (iPrPNP)Co(H)2(BPin) (iPrPNP = 2,6-(iPr2PCH2)2(C5H3N)), at both low and high substrate conversions. The overall first-order rate law and observation of a normal deuterium kinetic isotope effect on the borylation of benzofuran versus benzofuran-2-d1 support H2 reductive elimination from the cobalt(III) dihydride boryl as the turnover-limiting step. These findings stand in contrast to that established previously for the borylation of 2,6-lutidine with the same cobalt precatalyst, where borylation of the 4-position of the pincer occurred faster than the substrate turnover and arene C–H activation by a cobalt(I) boryl is turnover-limiting. Evaluation of the catalytic activity of different cobalt precursors in the C–H borylation of benzofuran with HBPin established that the ligand design principles for C–H borylation depend on the identities of both the arene and the boron reagent used: electron-donating groups improve catalytic activity of the borylation of pyridines and arenes with B2Pin2, whereas electron-withdrawing groups improve catalytic activity of the borylation of five-membered heteroarenes with HBPin. Catalyst deactivation by P–C bond cleavage from a cobalt(I) hydride was observed in the C–H borylation of arene substrates with C–H bonds that are less acidic than those of five-membered heteroarenes using HBPin and explains the requirement of B2Pin2 to achieve synthetically useful yields with these arene substrates. |  |
158 | Ammonia Activation, H2 Evolution and Nitride Formation from a Molybdenum Complex with a Chemically and Redox Noninnocent LigandGrant Margulieux, Máté J. Bezdek, Zoë R. Turner, and Paul J. Chirik J. Am. Chem. Soc. 2017, 139, 6110–6113. AbstractTreatment of the bis(imino)pyridine molybdenum η6-benzene complex (iPrPDI)Mo(η6-C6H6) (iPrPDI, 2,6-(2,6-iPr2C6H3N═CMe)2C5H3N) with NH3 resulted in coordination induced haptotropic rearrangement of the arene to form (iPrPDI)Mo(NH3)2(η2-C6H6). Analogous η2-ethylene and η2-cyclohexene complexes were also synthesized, and the latter was crystallographically characterized. All three compounds undergo loss of the η2-coordinated ligand followed by N–H bond activation, bis(imino)pyridine modification, and H2 loss. A dual ammonia activation approach has been discovered whereby reversible M–L cooperativity and coordination induced bond weakening likely contribute to dihydrogen formation. Significantly, the weakened N–H bonds in (iPrPDI)Mo(NH3)2(η2-C2H4) enabled hydrogen atom abstraction and synthesis of a terminal nitride from coordinated ammonia, a key step in NH3 oxidation. |  |
157 | Cobalt-Catalyzed 1,1-Diboration of Terminal Alkynes: Scope, Mechanism, and Synthetic ApplicationsSimon Krautwald, Máté J. Bezdek, and Paul J. Chirik J. Am. Chem. Soc. 2017, 139, 3868–3875. AbstractA cobalt-catalyzed method for the 1,1-diboration of terminal alkynes with bis(pinacolato)diboron (B2Pin2) is described. The reaction proceeds efficiently at 23 °C with excellent 1,1-selectivity and broad functional group tolerance. With the unsymmetrical diboron reagent PinB–BDan (Dan = naphthalene-1,8-diaminato), stereoselective 1,1-diboration provided products with two boron substituents that exhibit differential reactivity. One example prepared by diboration of 1-octyne was crystallized, and its stereochemistry established by X-ray crystallography. The utility and versatility of the 1,1-diborylalkene products was demonstrated in a number of synthetic applications, including a concise synthesis of the epilepsy medication tiagabine. In addition, a synthesis of 1,1,1-triborylalkanes was accomplished through cobalt-catalyzed hydroboration of 1,1-diborylalkenes with HBPin. Deuterium-labeling and stoichiometric experiments support a mechanism involving selective insertion of an alkynylboronate to a Co–B bond of a cobalt boryl complex to form a vinylcobalt intermediate. The latter was isolated and characterized by NMR spectroscopy and X-ray crystallography. A competition experiment established that the reaction involves formation of free alkynylboronate and the two boryl substituents are not necessarily derived from the same diboron source. |  |
156 | Benzyltriboronates: Building Blocks for Diastereoselective Carbon–Carbon Bond FormationW. Neil Palmer, Cayetana Zarate, and Paul J. Chirik J. Am. Chem. Soc. 2017, 139, 2589-2592. AbstractA highly diastereoselective carbon–carbon bond-forming reaction involving the tandem coupling of benzyltriboronates, enoates, and alkyl halides is described. This method was enabled by the discovery of α-diimine nickel catalysts that promote the chemoselective triborylation of benzylic C(sp3)–H bonds using B2Pin2 (Pin = pinacolate). The C–H functionalization method is effective with methylarenes and for the diborylation of secondary benzylic C–H bonds, providing direct access to polyboron building blocks from readily available hydrocarbons. Combination of the benzylic perborylation with a new deborylative conjugate addition–alkylation method enables a one-pot procedure in which multiple simple precursors are combined to generate diastereopure products containing quaternary stereocenters. |  |
155 | C(sp2)–H Borylation of Fluorinated Arenes Using an Air-Stable Cobalt Precatalyst: Electronically Enhanced Site Selectivity Enables Synthetic OpportunitiesJennifer V. Obligacion, Máté J. Bezdek, and Paul J. Chirik J. Am. Chem. Soc. 2017, 139, 2825-2832. AbstractCobalt catalysts with electronically enhanced site selectivity have been developed, as evidenced by the high ortho-to-fluorine selectivity observed in the C(sp2)–H borylation of fluorinated arenes. Both the air-sensitive cobalt(III) dihydride boryl 4-Me-(iPrPNP)Co(H)2BPin (1) and the air-stable cobalt(II) bis(pivalate) 4-Me-(iPrPNP)Co(O2CtBu)2 (2) compounds were effective and exhibited broad functional group tolerance across a wide range of fluoroarenes containing electronically diverse functional groups, regardless of the substitution pattern on the arene. The electronically enhanced ortho-to-fluorine selectivity observed with the cobalt catalysts was maintained in the presence of a benzylic dimethylamine and hydrosilanes, overriding the established directing-group effects observed with precious-metal catalysts. The synthetically useful selectivity observed with cobalt was applied to an efficient synthesis of the anti-inflammatory drug flurbiprofen. |  |
154 | Carbon-Carbon Bond Formation in a Weak Ligand Field: Leveraging Open Shell First Row Transition Metal CatalystsPaul J. Chirik Angew. Chem. Int. Ed. 2017, 56, 5170–5181. AbstractUnique features of earth-abundant transition-metal catalysts are reviewed in the context of catalytic carbon–carbon bond-forming reactions. Aryl-substituted bis(imino)pyridine iron and cobalt dihalide compounds, when activated with alkyl aluminum reagents, form highly active catalysts for the polymerization of ethylene. Open-shell iron and cobalt alkyl complexes have been synthesized that serve as single-component olefin polymerization catalysts. Reduced bis(imino)pyridine iron and cobalt dinitrogen compounds have also been discovered that promote the unique [2+2] cycloaddition of unactivated terminal alkenes. Studies of the electronic structure support open-shell intermediates, a deviation from traditional strong-field organometallic compounds that promote catalytic C−C bond formation. |  |
153 | Insight into Transmetalation Enables Cobalt-Catalyzed Suzuki–Miyaura Cross CouplingJamie M. Neely, Máté J. Bezdek, and Paul J. Chirik ACS Cent. Sci. 2016, 2, 935-942. AbstractAmong the fundamental transformations that comprise a catalytic cycle for cross coupling, transmetalation from the nucleophile to the metal catalyst is perhaps the least understood. Optimizing this elementary step has enabled the first example of a cobalt-catalyzed Suzuki–Miyaura cross coupling between aryl triflate electrophiles and heteroaryl boron nucleophiles. Key to this discovery was the preparation and characterization of a new class of tetrahedral, high-spin bis(phosphino)pyridine cobalt(I) alkoxide and aryloxide complexes, (iPrPNP)CoOR, and optimizing their reactivity with 2-benzofuranylBPin (Pin = pinacolate). Cobalt compounds with small alkoxide substituents such as R = methyl and ethyl underwent swift transmetalation at 23 °C but also proved kinetically unstable toward β–H elimination. Secondary alkoxides such as R = iPr or CH(Ph)Me balanced stability and reactivity. Isolation and structural characterization of the product following transmetalation, (iPrPNP)Co(2-benzofuranyl), established a planar, diamagnetic cobalt(I) complex, demonstrating the high- and low-spin states of cobalt(I) rapidly interconvert during this reaction. The insights from the studies in this elementary step guided selection of appropriate reaction conditions to enable the first examples of cobalt-catalyzed C–C bond formation between neutral boron nucleophiles and aryl triflate electrophiles, and a model for the successful transmetalation reactivity is proposed. | 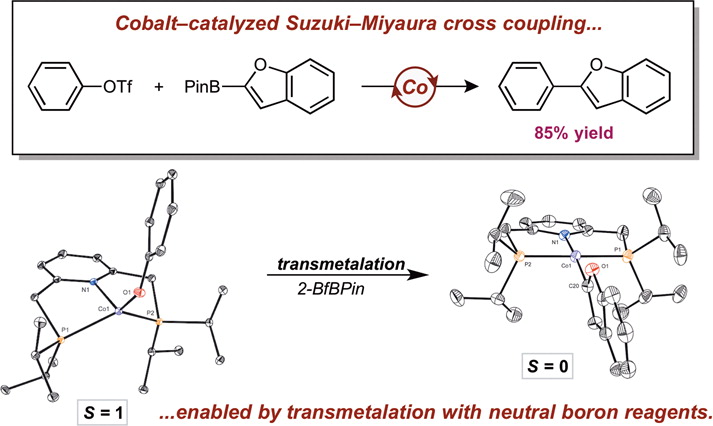 |
152 | Coordination-induced weakening of ammonia, water, and hydrazine X–H bonds in a molybdenum complexMáté J. Bezdek, Sheng Guo and Paul J. Chirik AbstractAlthough scores of transition metal complexes incorporating ammonia or water ligands have been characterized over the past century, little is known about how coordination influences the strength of the nitrogen-hydrogen and oxygen-hydrogen bonds. Here we report the synthesis of a molybdenum ammonia complex supported by terpyridine and phosphine ligands that lowers the nitrogen-hydrogen bond dissociation free energy from 99.5 (gas phase) to an experimentally measured value of 45.8 kilocalories per mole (agreeing closely with a value of 45.1 kilocalories per mole calculated by density functional theory). This bond weakening enables spontaneous dihydrogen evolution upon gentle heating, as well as the hydrogenation of styrene. Analogous molybdenum complexes promote dihydrogen evolution from coordinated water and hydrazine. Electrochemical and theoretical studies elucidate the contributions of metal redox potential and ammonia acidity to this effect. | |
151 | Cobalt-Catalyzed C(sp2)–H Borylation with an Air-Stable, Readily Prepared Terpyridine Cobalt(II) Bis(acetate) PrecatalystNadia G. Léonard, Máté J. Bezdek, and Paul J. Chirik Organometallics 2017, 36, 142-150. AbstractA bench-stable, 4-aryl-substituted terpyridine supported, high-spin cobalt(II) bis(acetate) complex, (ArTpy)Co(OAc)2 (ArTpy = 4′-(4-N,N′-dimethylaminophenyl)-2,2′:6′,2″-terpyridine), is active for the C(sp2)–H borylation of arenes and heteroarenes with B2Pin2 (Pin = pinacolato). Optimization of the catalytic borylation reaction revealed improved performance in the presence of LiOMe and turnover numbers of up to 100 have been observed using all air-stable components. EPR specstroscopy identified formation of inactive cobalt species, promoted by excess HBPin. A high-spin cobalt(II) bis[(diacetoxy)pinacolatoborate−κ3O,O,O] compound has been isolated and characterized by X-ray diffraction and is the result of catalyst deactivation. | 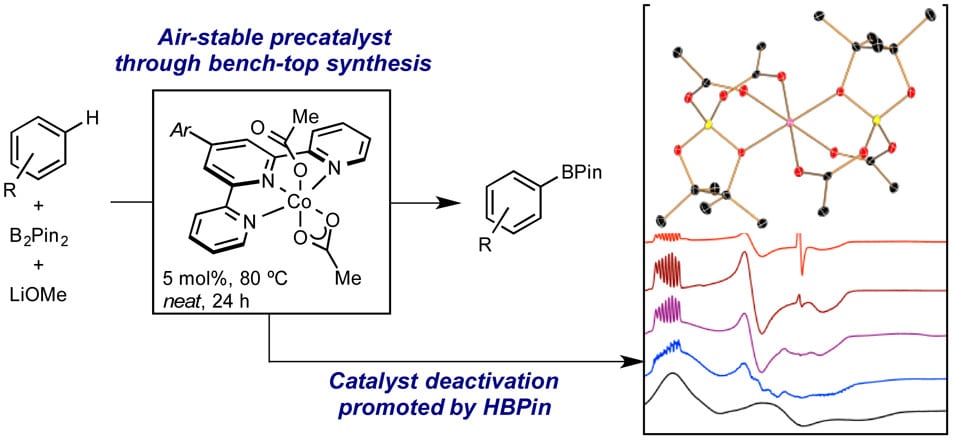 |
150 | Catalytic Proton Coupled Electron Transfer from Metal Hydrides to Titanocene Amides, Hydrazides and Imides: Determination of Thermodynamic Parameters Relevant to Nitrogen FixationIraklis Pappas and Paul J. Chirik J. Am. Chem. Soc. 2016, 138, 13379-13389. AbstractThe hydrogenolysis of titanium–nitrogen bonds in a series of bis(cyclopentadienyl) titanium amides, hydrazides and imides by proton coupled electron transfer (PCET) is described. Twelve different N–H bond dissociation free energies (BDFEs) among the various nitrogen-containing ligands were measured or calculated, and effects of metal oxidation state and N-ligand substituent were determined. Two metal hydride complexes, (η5-C5Me5)(py-Ph)Rh–H (py-Ph = 2-pyridylphenyl, [Rh]-H) and (η5-C5R5)(CO)3Cr−H ([Cr]R-H, R= H, Me) were evaluated for formal H atom transfer reactivity and were selected due to their relatively weak M–H bond strengths yet ability to activate and cleave molecular hydrogen. Despite comparable M–H BDFEs, disparate reactivity between the two compounds was observed and was traced to the vastly different acidities of the M–H bonds and overall redox potentials of the molecules. With [Rh]–H, catalytic syntheses of ammonia, silylamine and N,N-dimethylhydrazine have been accomplished from the corresponding titanium(IV) complex using H2 as the stoichiometric H atom source. The data presented in this study provides the thermochemical foundation for the synthesis of NH3 by proton coupled electron transfer at a well-defined transition metal center. |  |
149 | Cationic Pyridine(diimine) Iron Tethered Alkene Complexes: Synthetic Models For Elusive Intermediates In Iron-Catalyzed Ethylene PolymerizationBrian A. Schaefer, Grant W. Margulieux, and Paul J. Chirik Bull. Jpn. Soc. Coord. Chem. 2016, 67, 19-29. AbstractCationic bis(imino)pyridine iron alkene complexes, key intermediates in the polymerization and oligomerization of ethylene, have been targeted to understand important physical properties such as spin state of the metal and the role of the redox-active supporting ligand. The allyl derivative, (MePDI)Fe(h3-C3H5) was synthesized but proved unstable toward one electron oxidation with ferrocenium reagents. Tethered alkoxide-alkene complexes were explored as more well behaved alternatives and (MePDI) Fe(OC(Ph)2CH2(h2-CHCH2) was synthesized, crystallographically characterized and determined to be a high spin Fe(II) complex with a bis(imino)pyridine radical anion. One electron oxidation with [Cp2Fe][BArF24] (BArF24 = C6H3-3,5-(CF3)2) generated the desired cationic iron complex, [(MePDI)Fe(OC(Ph)2(h2-C3H5))][BArF24]. The solid-state structure confi rmed alkene coordination and provided the metrical parameters used to establish a neutral bis(imino)pyridine chelate. The observation of a high spin Fe(II) complex demonstrates that the iron does not change spin state upon olefi n coordination. | 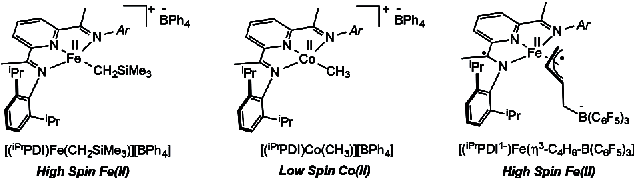 |
148 | Cobalt-Catalyzed C(sp2)-H Borylation: Mechanistic Insights Inspire Catalyst DesignJennifer V. Obligacion, Scott P. Semproni, Iraklis Pappas and Paul J. Chirik J. Am. Chem. Soc. 2016, 138, 10645-10653. AbstractA comprehensive study into the mechanism of bis(phosphino)pyridine (PNP) cobalt-catalyzed C–H borylation of 2,6-lutidine using B2Pin2 (Pin = pinacolate) has been conducted. The experimentally observed rate law, deuterium kinetic isotope effects, and identification of the catalyst resting state support turnover limiting C–H activation from a fully characterized cobalt(I) boryl intermediate. Monitoring the catalytic reaction as a function of time revealed that borylation of the 4-position of the pincer in the cobalt catalyst was faster than arene borylation. Cyclic voltammetry established the electron withdrawing influence of 4-BPin, which slows the rate of C–H oxidative addition and hence overall catalytic turnover. This mechanistic insight inspired the next generation of 4-substituted PNP cobalt catalysts with electron donating and sterically blocking methyl and pyrrolidinyl substituents that exhibited increased activity for the C–H borylation of unactivated arenes. The rationally designed catalysts promote effective turnover with stoichiometric quantities of arene substrate and B2Pin2. Kinetic studies on the improved catalyst, 4-(H)2BPin, established a change in turnover limiting step from C–H oxidative addition to C–B reductive elimination. The iridium congener of the optimized cobalt catalyst, 6-(H)2BPin, was prepared and crystallographically characterized and proved inactive for C–H borylation, a result of the high kinetic barrier for reductive elimination from octahedral Ir(III) complexes. | 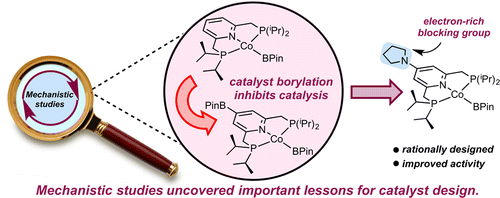 |
147 | Thermodynamics of N–H bond formation in bis(phosphine) molybdenum(II) diazenides and the influence of the trans ligandMáté J. Bezdek and Paul J. Chirik Dalton Trans. 2016, 45, 15922-15930. AbstractA series of bis(phosphine) molybdenum(II) diazenides [(dppe)2Mo(NNCy)(I)], [(dppe)2(CH3CN)Mo(NNCy)][BArF24] and [(dppe)2)(3,5-(CF3)2C6H3CN)Mo(NNCy)][BArF24] (dppe = 1,2-bis(diphenylphosphino)ethane; Cy = cyclohexyl; ArF24 = (3,5-(CF3)2C6H3)4) were synthesized and structurally characterized. Treatment of the diazenido complexes with a stoichiometric amount of [H(OEt2)2][BArF24] afforded the corresponding molybdenum(IV) hydrazido species [(dppe)2Mo(NNHCy)(I)][BArF24], [(dppe)2(CH3CN)Mo(NNHCy)][BArF24]2 and [(dppe)2(3,5-(CF3)2C6H3CN)Mo(NNHCy)][BArF24]2, enabling the study of N–H bond dissociation free energies (BDFEs) in the classical Chatt-type bis(phosphine) diazenide platform as a function of ligand (L) trans to the nitrogenous fragment. Deprotonation and electrochemical experiments established that the trans nitrile 3,5-(CF3)2C6H3CN afforded the least reducing molybdenum(IV) hydrazido complex in the series ( |  |
146 | Expanding Boundaries: N2 Cleavage and Functionalization beyond Early Transition MetalsMáté J. Bezdek and Paul J. Chirik Angew. Chem. Int. Ed. 2016, 55, 7892-7896 AbstractEarly transition metals are well known to catalyze the cleavage and functionalization of N2. In this Highlight, recent work showing that a rhenium catalyst is also capable of carrying out this difficult task is summarized, and a synthetic cycle for the stoichiometric incorporation of atmospheric N2 into acetonitrile is presented. | 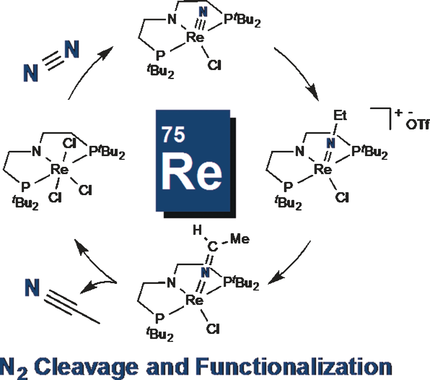 |
145 | Alkene Hydrosilylation Using Tertiary Silanes with α-Diimine Nickel Catalysts. Redox-Active Ligands Promote a Distinct Mechanistic Pathway from Platinum CatalystsIraklis Pappas, Sean Treacy, and Paul J. Chirik ACS Catal. 2016, 6, 4105-4109. AbstractCombination of the readily available α-diimine ligand, ((ArN═C(Me))2 Ar = 2,6-iPr2–C6H3), (iPrDI) with air-stable nickel(II) bis(carboxylates) generated a highly active catalyst exhibiting anti-Markovnikov selectivity for the hydrosilylation of alkenes with a variety of industrially relevant tertiary alkoxy- and siloxy-substituted silanes. A combination of the method of continuous variations with stoichiometric studies identified the formally Ni(I) hydride dimer, [(iPrDI)NiH]2 as the nickel compound formed following reduction of the carboxylate ligands. For the hydrosilylation of 1-octene with (EtO)3SiH, a rate law of [Ni]1/2[1-octene][(EtO)3SiH] in combination with deuterium-labeling studies establish dissociation of the nickel hydride dimer followed by fast and reversible alkene insertion into (iPrDI)NiH, consistent with turnover-limiting C–Si bond formation. The hydrosilylation of 1-octene with triethoxysilane, a reaction performed commercially in the silicones industry on a scale of >5 000 000 kg/year, was conducted on a 10 g scale with 96% yield and >98% selectivity for the desired product. Silicone cross-linking, another major industrial application of homogeneous hydrosilylation, was also demonstrated using the air-stable nickel and ligand precursors. |  |
144 | Bench-Stable, Substrate-Activated Cobalt Carboxylate Pre-Catalysts for Alkene Hydrosilylation with Tertiary SilanesChristopher H. Schuster, Tianning Diao, Iraklis Pappas, and Paul J. Chirik ACS Catal. 2016, 6, 2632-2636. AbstractHigh-spin pyridine diimine cobalt(II) bis(carboxylate) complexes have been synthesized and exhibit high activity for the hydrosilylation of a range of commercially relevant alkenes and tertiary silanes. Previously observed dehydrogenative silylation is suppressed with the use of sterically unencumbered ligands, affording exclusive hydrosilylation with up to 4000 TON. The cobalt precatalysts were readily prepared and handled on the benchtop and underwent substrate activation, obviating the need for external reductants. The cobalt catalysts are tolerant of epoxide, amino, carbonyl, and alkyl halide functional groups, broadening the scope of alkene hydrosilylation with earth-abundant metal catalysts. |  |
143 | Terpyridine Molybdenum Dinitrogen Chemistry: Synthesis of Dinitrogen Complexes That Vary by Five Oxidation StatesMáté J. Bezdek, Sheng Guo, and Paul J. Chirik Inorg. Chem. 2016, 55, 3117-3127. AbstractA bimetallic molybdenum complex bridged by an activated dinitrogen ligand and supported by phosphine and terpyridine ligands, [{(PhTpy)(PPh2Me)2Mo}2(μ2-N2)][BArF24]2 [PhTpy = 4′-Ph-2,2′,6′,2″-terpyridine; ArF24 = (C6H3-3,5-(CF3)2)4], was synthesized and structurally characterized, and its electronic structure was determined using a combination of experimental and density functional theory computational methods. Each molybdenum atom is best described as molybdenum(II) bridged by a modestly activated [N2]2– ligand. The cyclic voltammogram of [{(PhTpy)(PPh2Me)2Mo}2(μ2-N2)]2+ displays two reversible reductive and two reversible oxidative features, prompting the preparation and characterization of a series of molybdenum dinitrogen compounds spanning five oxidation states ([{(PhTpy)(PPh2Me)2Mo}2(μ2-N2)][BArF24]n, where n = 4, 3, 2, 1, 0). Raman and 15N NMR spectroscopic data establish that the bridging nitrogen ligand remains intact across the redox series. Electron paramagnetic resonance spectroscopy was used to probe the nature of the unpaired electron in the mixed-valent electronic oxidized and reduced products. The singly occupied molecular orbital is principally metal-based in [{(PhTpy)(PPh2Me)2Mo}2(μ2-N2)]3+ and ligand-localized in [{(PhTpy)(PPh2Me)2Mo}2(μ2-N2)]+. | 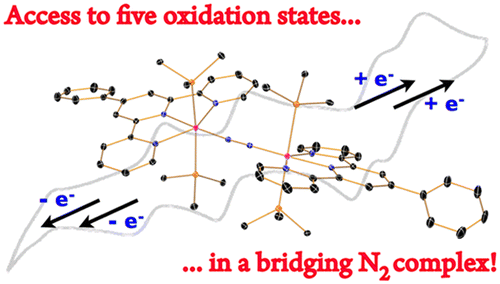 |
142 | Nickel-Catalyzed Asymmetric Alkene Hydrogenation of α,β-Unsaturated Esters: High-Throughput Experimentation-Enabled Reaction Discovery, Optimization, and Mechanistic ElucidationMichael Shevlin, Max R. Friedfeld, Huaming Sheng, Nicholas A. Pierson, Jordan M. Hoyt, Louis-Charles Campeau, and Paul J. Chirik J. Am. Chem. Soc. 2016, 138, 3562-3569. AbstractA highly active and enantioselective phosphine-nickel catalyst for the asymmetric hydrogenation of α,β-unsaturated esters has been discovered. The coordination chemistry and catalytic behavior of nickel halide, acetate, and mixed halide-acetate with chiral bidentate phosphines have been explored and deuterium labeling studies, the method of continuous variation, nonlinear studies, and kinetic measurements have provided mechanistic understanding. Activation of molecular hydrogen by a trimeric (Me–DuPhos)3Ni3(OAc)5I complex was established as turnover limiting followed by rapid conjugate addition of a nickel hydride and nonselective protonation to release the substrate. In addition to reaction discovery and optimization, the previously unreported utility high-throughput experimentation for mechanistic elucidation is also described. | 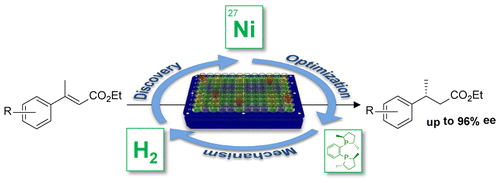 |
141 | Cobalt-Catalyzed Enantioselective Hydrogenation of Minimally Functionalized Alkenes: Isotopic Labeling Provides Insight into the Origin of Stereoselectivity and Alkene Insertion PreferencesMax R. Friedfeld, Michael Shevlin, Grant W. Margulieux, Louis-Charles Campeau, and Paul J. Chirik J. Am. Chem. Soc. 2016, 138, 3314-3324. AbstractThe asymmetric hydrogenation of cyclic alkenes lacking coordinating functionality with a C1-symmetric bis(imino)pyridine cobalt catalyst is described and has been applied to the synthesis of important substructures found in natural products and biologically active compounds. High activities and enantioselectivities were observed with substituted benzo-fused five-, six-, and seven-membered alkenes. The stereochemical outcome was dependent on both the ring size and exo/endo disposition. Deuterium labeling experiments support rapid and reversible 2,1-insertion that is unproductive for generating alkane product but accounts for the unusual isotopic distribution in deuterated alkanes. Analysis of the stereochemical outcome of the hydrogenated products coupled with isotopic labeling, stoichiometric, and kinetic studies established 1,2-alkene insertion as both turnover limiting and enantiodetermining with no evidence for erosion of cobalt alkyl stereochemistry by competing β-hydrogen elimination processes. A stereochemical model accounting for the preferred antipodes of the alkanes is proposed and relies on the subtle influence of the achiral aryl imine substituent on the cobalt catalyst. | 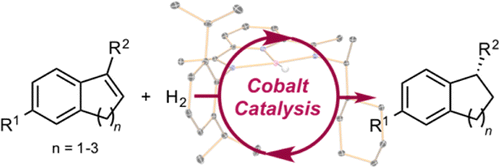 |
140 | Cobalt-Catalyzed Benzylic Borylation: Enabling Polyborylation and Functionalization of Remote, Unactivated C(sp3)–H BondsW. Neil Palmer, Jennifer V. Obligacion, Iraklis Pappas, and Paul J. Chirik J. Am. Chem. Soc. 2016, 138, 766-769. AbstractCobalt dialkyl and bis(carboxylate) complexes bearing α-diimine ligands have been synthesized and demonstrated as active for the C(sp3)-H borylation of a range of substituted alkyl arenes using B2Pin2 (Pin = pinacolate) as the boron source. At longer reaction times, rare examples of polyborylation were observed, and in the case of toluene, all three benzylic C-H positions were functionalized. Coupling benzylic C-H activation with alkyl isomerization enabled a base-metal-catalyzed method for the borylation of remote, unactivated C(sp3)-H bonds. | 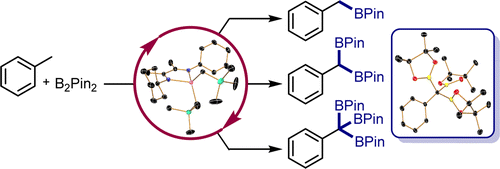 |
139 | Iron-catalysed tritiation of pharmaceuticalsRenyuan Pony Yu, David Hesk, Nelo Rivera, István Pelczer, and Paul J. Chirik AbstractA thorough understanding of the pharmacokinetic and pharmacodynamic properties of a drug in animal models is a critical component of drug discovery and development1,2,3,4,5,6. Such studies are performed in vivo and in vitro at various stages of the development process—ranging from preclinical absorption, distribution, metabolism and excretion (ADME) studies to late-stage human clinical trials—to elucidate a drug molecule’s metabolic profile and to assess its toxicity2. Radiolabelled compounds, typically those that contain 14C or 3H isotopes, are one of the most powerful and widely deployed diagnostics for these studies4,5. The introduction of radiolabels using synthetic chemistry enables the direct tracing of the drug molecule without substantially altering its structure or function. The ubiquity of C–H bonds in drugs and the relative ease and low cost associated with tritium (3H) make it an ideal radioisotope with which to conduct ADME studies early in the drug development process2,4,6. Here we describe an iron-catalysed method for the direct 3H labelling of pharmaceuticals by hydrogen isotope exchange, using tritium gas as the source of the radioisotope. The site selectivity of the iron catalyst is orthogonal to currently used iridium catalysts and allows isotopic labelling of complementary positions in drug molecules, providing a new diagnostic tool in drug development. | |
138 | Synthesis and Electronic Structure of Iron Borate Betaine Complexes as a Route to Single-Component Iron Ethylene Oligomerization and Polymerization CatalystsBrian A. Schaefer, Grant W. Margulieux, Margaret A. Tiedmann, Brooke L. Small, and Paul J. Chirik Organometallics 2015, 34, 5615-5623. AbstractA new route to single-component iron ethylene oligomerization and polymerization catalysts is described. Treatment of readily synthesized iron butadiene complexes with B(C6F5)3 generated the corresponding betaine compounds, active catalysts for the oligomerization and polymerization of ethylene. The electronic structures of a family of iron compounds bearing tridentate, α-diimine phosphine ligands have been determined, including cases where the neutral donor has dissociated from the metal. In iron-catalyzed ethylene oligomerization with these compounds, the hemilability of the chelate has been identified as a catalyst deactiviation pathway. |  |
137 | Iron-catalyzed intermolecular [2+2] cycloadditions of unactivated alkenesJordan M. Hoyt, Valerie A. Schmidt, Aaron M. Tondreau, and Paul J. Chirik AbstractCycloadditions, such as the [4+2] Diels-Alder reaction to form six-membered rings, are among the most powerful and widely used methods in synthetic chemistry. The analogous [2+2] alkene cycloaddition to synthesize cyclobutanes is kinetically accessible by photochemical methods, but the substrate scope and functional group tolerance are limited. Here, we report iron-catalyzed intermolecular [2+2] cycloaddition of unactivated alkenes and cross cycloaddition of alkenes and dienes as regio- and stereoselective routes to cyclobutanes. Through rational ligand design, development of this base metal–catalyzed method expands the chemical space accessible from abundant hydrocarbon feedstocks. | ![Iron-catalyzed intermolecular [2+2] cycloadditions of unactivated alkenes](/wp-content/uploads/2022/10/img-publ-137.png) |
136 | Iron- and Cobalt-Catalyzed Alkene Hydrogenation: Catalysis with Both Redox-Active and Strong Field LigandsPaul J. Chirik Acc. Chem. Res. 2015, 48, 1687–1695. AbstractThe hydrogenation of alkenes is one of the most impactful reactions catalyzed by homogeneous transition metal complexes finding application in the pharmaceutical, agrochemical, and commodity chemical industries. For decades, catalyst technology has relied on precious metal catalysts supported by strong field ligands to enable highly predictable two-electron redox chemistry that constitutes key bond breaking and forming steps during turnover. Alternative catalysts based on earth abundant transition metals such as iron and cobalt not only offer potential environmental and economic advantages but also provide an opportunity to explore catalysis in a new chemical space. The kinetically and thermodynamically accessible oxidation and spin states may enable new mechanistic pathways, unique substrate scope, or altogether new reactivity. This Account describes my group’s efforts over the past decade to develop iron and cobalt catalysts for alkene hydrogenation. Particular emphasis is devoted to the interplay of the electronic structure of the base metal compounds and their catalytic performance. First generation, aryl-substituted pyridine(diimine) iron dinitrogen catalysts exhibited high turnover frequencies at low catalyst loadings and hydrogen pressures for the hydrogenation of unactivated terminal and disubstituted alkenes. Exploration of structure–reactivity relationships established smaller aryl substituents and more electron donating ligands resulted in improved performance. Second generation iron and cobalt catalysts where the imine donors were replaced by N-heterocyclic carbenes resulted in dramatically improved activity and enabled hydrogenation of more challenging unactivated, tri- and tetrasubstituted alkenes. Optimized cobalt catalysts have been discovered that are among the most active homogeneous hydrogenation catalysts known. Synthesis of enantiopure, C1 symmetric pyridine(diimine) cobalt complexes have enabled rare examples of highly enantioselective hydrogenation of a family of substituted styrene derivatives. Because improved hydrogenation performance was observed with more electron rich supporting ligands, phosphine cobalt(II) dialkyl complexes were synthesized and found to be active for the diastereoselective hydrogenation of various substituted alkenes. Notably, this class of catalysts was activated by hydroxyl functionality, representing a significant advance in the functional group tolerance of base metal hydrogenation catalysts. Through collaboration with Merck, enantioselective variants of these catalysts were discovered by high throughput experimentation. Catalysts for the hydrogenation of functionalized and essentially unfunctionalized alkenes have been discovered using this approach. Development of reliable, readily accessible cobalt precursors facilitated catalyst discovery and may, along with lessons learned from electronic structure studies, provide fundamental design principles for catalysis with earth abundant transition metals beyond alkene hydrogenation. |  |
135 | Alkene Isomerization–Hydroboration Promoted by Phosphine-Ligated Cobalt CatalystsMargaret L. Scheuermann, Elizabeth J. Johnson, and Paul J. Chirik Org. Lett. 2015, 17, 2716-2719. AbstractGenerated in situ from air-stable cobalt precursors or readily synthesized using NaHBEt3, (PPh3)3CoH(N2) was found to be an effective catalyst for the hydroboration of alkenes. Unlike previous base-metal catalysts for alkene isomerization–hydroboration which favor the incorporation of boron at terminal positions, (PPh3)3CoH(N2) promotes boron incorporation adjacent to π-systems even in substrates where the alkene is at a remote position, enabling a unique route to 1,1-diboron compounds from α,ω-dienes. | 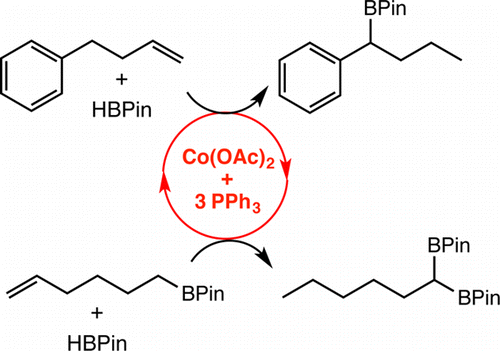 |
134 | Cobalt-Catalyzed [2π + 2π] Cycloadditions of Alkenes: Scope, Mechanism, and Elucidation of Electronic Structure of Catalytic IntermediatesValerie A. Schmidt, Jordan M. Hoyt, Grant M. Margulieux, and Paul J. Chirik J. Am. Chem. Soc. 2015, 137, 7903-7914. AbstractAryl-substituted bis(imino)pyridine cobalt dinitrogen compounds, (RPDI)CoN2, are effective precatalysts for the intramolecular [2π + 2π] cycloaddition of α,ω-dienes to yield the corresponding bicyclo[3.2.0]heptane derivatives. The reactions proceed under mild thermal conditions with unactivated alkenes, tolerating both amine and ether functional groups. The overall second order rate law for the reaction, first order with respect to both the cobalt precatalyst and the substrate, in combination with electron paramagnetic resonance (EPR) spectroscopic studies established the catalyst resting state as dependent on the identity of the precatalyst and diene substrate. Planar S =1/2 κ3-bis(imino)pyridine cobalt alkene and tetrahedral κ2-bis(imino)pyridine cobalt diene complexes were observed by EPR spectroscopy and in the latter case structurally characterized. The hemilabile chelate facilitates conversion of a principally ligand-based singly occupied molecular orbital (SOMO) in the cobalt dinitrogen and alkene compounds to a metal-based SOMO in the diene intermediates, promoting C–C bond-forming oxidative cyclization. Structure–activity relationships on bis(imino)pyridine substitution were also established with 2,4,6-tricyclopentyl-substituted aryl groups, resulting in optimized catalytic [2π + 2π] cycloaddition. The cyclopentyl groups provide a sufficiently open metal coordination sphere that encourages substrate coordination while remaining large enough to promote a challenging, turnover-limiting C(sp3)–C(sp3) reductive elimination. | ![Cobalt-Catalyzed [2π + 2π] Cycloadditions of Alkenes: Scope, Mechanism, and Elucidation of Electronic Structure of Catalytic Intermediates](/wp-content/uploads/2022/10/img-publ-134.gif) |
133 | Cobalt Catalyzed Z-Selective Hydroboration of Terminal Alkynes and Elucidation of the Origin of SelectivityJennifer V. Obligacion, Jamie M. Neely, Aliza N. Yazdani, Iraklis Pappas, and Paul J. Chirik J. Am. Chem. Soc. 2015, 137, 5855-5858. AbstractA bis(imino)pyridine cobalt-catalyzed hydroboration of terminal alkynes with HBPin (Pin = pinacolate) with high yield and (Z)-selectivity for synthetically valuable vinylboronate esters is described. Deuterium labeling studies, stoichiometric experiments, and isolation of catalytically relevant intermediates support a mechanism involving selective insertion of an alkynylboronate ester into a Co–H bond, a pathway distinct from known precious metal catalysts where metal vinylidene intermediates have been proposed to account for the observed (Z) selectivity. The identity of the imine substituents dictates the relative rates of activation of the cobalt precatalyst with HBPin or the terminal alkyne and, as a consequence, is responsible for the stereochemical outcome of the catalytic reaction. | 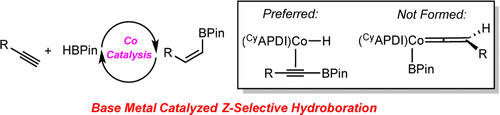 |
132 | Evaluation of Cobalt Complexes Bearing Tridentate Pincer Ligands for Catalytic C–H BorylationBrian A. Schaefer, Grant W. Margulieux, Brooke L. Small, and Paul J. Chirik Organometallics 2015, 34, 1307-1320. AbstractCobalt(II) dichloride complexes supported by a variety of neutral, tridentate pincer ligands have been prepared and, following in situ activation with NaBEt3H, evaluated for the catalytic borylation of 2-methylfuran, 2,6-lutidine, and benzene using both HBPin and B2Pin2 (Pin = pinacolate) as boron sources. Preparation of well-defined organometallic compounds in combination with stoichiometric experiments with HBPin and B2Pin2 provided insight into the nature and kinetic stability of the catalytically relevant species. In cases where sufficiently electron donating pincers are present, such as with bis(phosphino)pyridine chelates, Co(III) resting states are preferred and catalytic C–H borylation is efficient. Introduction of a redox-active subunit into the pincer reduces its donating ability and, as a consequence, the accessibility of a Co(III) resting state. In these cases, unusual mixed-valent μ-hydride cobalt complexes have been crystallographically and spectroscopically characterized. These studies have also shed light on the active species formed during in situ activated cobalt alkene hydroboration catalysis and provide important design criteria in base metal catalyzed C–B bond forming reactions. |  |
131 | Ammonia Synthesis by Hydrogenolysis of Titanium–Nitrogen Bonds Using Proton Coupled Electron TransferIraklis Pappas and Paul J. Chirik J. Am. Chem. Soc. 2015, 137, 3498-3501. AbstractThe catalytic hydrogenolysis of the titanium–amide bond in (η5-C5Me4SiMe3)2Ti(Cl)NH2 to yield free ammonia is described. The rhodium hydride, (η5-C5Me5)(py-Ph)RhH (py-Ph = 2-phenylpyridine), serves as the catalyst and promotes N–H bond formation via hydrogen atom transfer. The N–H bond dissociation free energies of ammonia ligands have also been determined for titanocene and zirconocene complexes and reveal a stark dependence on metal identity and oxidation state. In all cases, the N–H BDFEs of coordinated NH3 decreases by >40 kcal/mol from the value in the free gas phase molecule. |  |
130 | A Career in Catalysis: John E. BercawPeter T. Wolczanski and Paul J. Chirik ACS Catal. 2015, 5, 1747-1757. AbstractOn the occasion of Professor John Bercaw’s 70th birthday, we reflect and highlight his distinguished career in organometallic chemistry and homogeneous catalysis. What began as a fundamental interest in the chemistry of bis(cyclopentadienyl)titanium compounds and their interaction with molecular nitrogen evolved into a vibrant and diverse program tackling some of the most important problems in catalysis. Using well-defined organometallic compounds, fundamental insights were gained in the mechanism of CO reduction; basic transformations of organometallic chemistry, such as alkene insertion and alkyl β-hydrogen elimination; the origin of stereocontrol in metallocene-catalyzed polymerization; and in the activation of hydrocarbons by electrophilic late transition metals. |  |
129 | High-Activity Cobalt Catalysts for Alkene Hydroboration with Electronically Responsive Terpyridine and α-Diimine LigandsW. Neil Palmer, Tianning Diao, Iraklis Pappas and Paul J. Chirik AbstractCobalt alkyl complexes bearing readily available and redox-active 2,2′:6′,2″-terpyridine and α-diimine ligands have been synthesized, and their electronic structures have been elucidated. In each case, the supporting chelate is reduced to the monoanionic, radical form that is engaged in antiferromagnetic coupling with the cobalt(II) center. Both classes of cobalt alkyls proved to be effective for the isomerization–hydroboration of sterically hindered alkenes. An α-diimine-substituted cobalt allyl complex proved exceptionally active for the reduction of hindered tri-, tetra-, and geminally substituted alkenes, representing one of the most active homogeneous catalysts known for hydroboration. With limonene, formation of an η3-allyl complex with a C–H agostic interaction was identified and accounts for the sluggish reactivity observed with diene substrates. For the terpyridine derivative, unique Markovnikov selectivity with styrene was also observed with HBPin. | 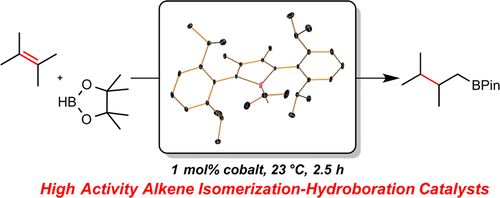 |
128 | Synthesis and carbonylation of oxotitanocyclobutanes derived from allene cycloaddition to a monomeric titanocene oxide.Pappas, I.; Chirik, P. J. | |
127 | Synthesis and Ligand Modification Chemistry of a Molybdenum Dinitrogen Complex: Redox and Chemical Activity of a Bis(imino)pyridine Ligand.Margulieux, G. W.; Turner, Z. R.; Chirik, P. J. | |
126 | Electronic Structure Determination of Pyridine N-Heterocyclic Carbene Iron Dinitrogen Complexes and Neutral Ligand Derivatives.Darmon, J. M.; Yu, R. P.; Semproni, S. P.; Turner, Z. R.; Stieber, S. C. E.; DeBeer, S.; Chirik, P. J. | 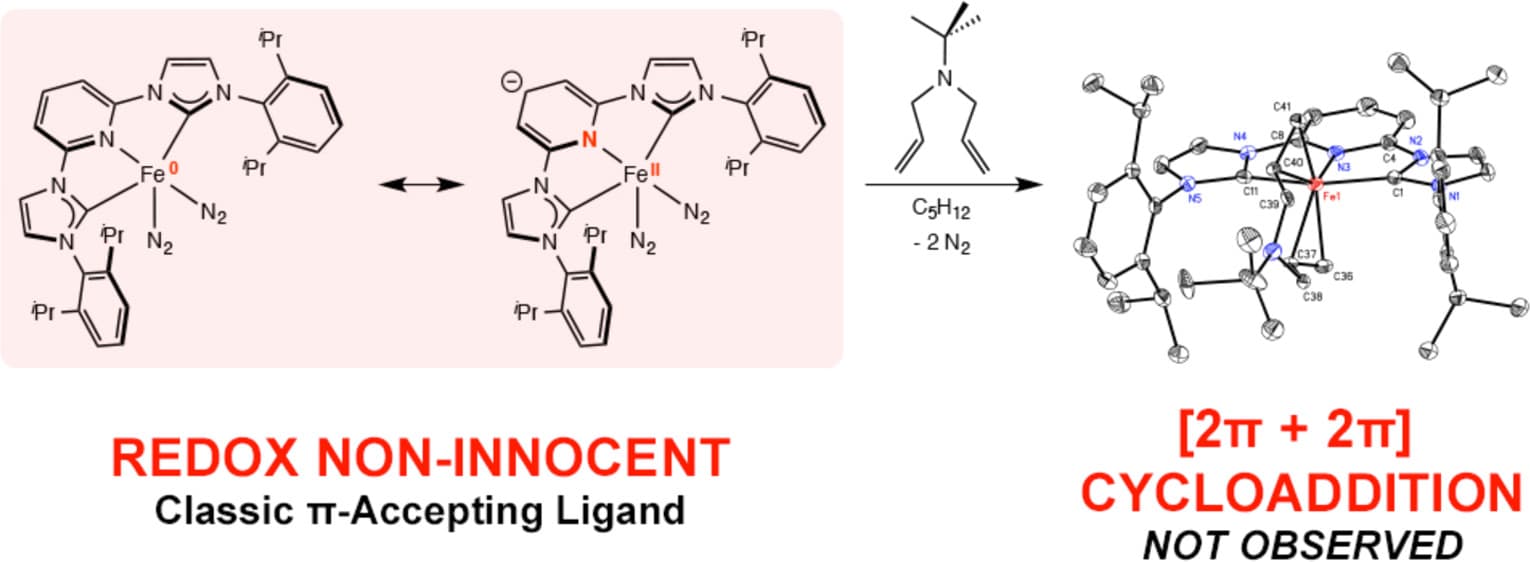 |
125 | Bis(phosphine)cobalt Dialkyl Complexes for Directed Catalytic Alkene Hydrogenation.Friedfeld, M. R.; Margulieux, G. W.; Schaefer, B. A.; Chirik, P. J. | |
124 | Carbon Dioxide Hydrosilylation Promoted by Cobalt Pincer Complexes.Scheuermann, M. L.; Semproni, S. P.; Pappas, I.; Chirik, P. J. | |
123 | Bis(imino)pyridine Cobalt-Catalyzed Dehydrogenative Silylation of Alkenes: Scope, Mechanism, and Origins of Selective Allylsilane Formation.Atienza, C. C. H.; Diao, T.; Weller, K. J.; Nye, S. A.; Lewis, K. M.; Delis, J. G. P.; Boyer, J. L.; Roy, A. K.; Chirik, P. J. | |
122 | N–N Bond Cleavage of 1,2-Diarylhydrazines and N–H Bond Formation via H-Atom Transfer in Vanadium Complexes Supported by a Redox- Active Ligand.Milsmann, C.; Semproni, S. P.; Chirik, P. J. | |
121 | N–H and N–C Bond Formation with an N2-Derived Dihafnium μ- Nitrido Complex.Semproni, S. P.; Chirik, P. J. | |
120 | Four-Coordinate Cobalt Pincer Complexes: Electronic Structure Studies and Ligand Modification by Homolytic and Heterolytic Pathways.Semproni, S. P.; Milsmann, C.; Chirik, P. J. | |
119 | Synthesis and Hydrogenation Activity of Iron Dialkyl Complexes with Chiral Bidentate Phosphines.Hoyt, J. M.; Shevlin, M.; Margulieux, G. W.; Krska, S. W.; Tudge, M. T.; Chirik, P. J. | |
118 | Alkyne Cycloaddition to a Titanocene Oxide as a Route to Cyclopentadienyl Modification.Pappas, I.; Chirik, P. J. | |
117 | Photochemically Induced Reductive Elimination as a Route to a Zirconocene Complex with a Strongly Activated N2 Ligand.Margulieux, G. M.; Semproni, S. P.; Chirik, P. J. | |
116 | Cobalt-Catalyzed C–H Borylation.Obligacion, J. V.; Semproni, S. P.; Chirik, P. J. | |
115 | Oxidative addition and C–H activation chemistry with a PNP pincer-ligated cobalt complex.Semproni, S. P.; Atienze, C. C. H.; Chirik, P. J. | |
114 | Synthesis, electronic structure and reactivity of bis(imino)pyridine iron carbene complexes: evidence for a carbene radical.Russel, S. K.; Hoyt, J. M.; Bart, S. C.; Milsmann, C.; Stieber, S. C. E.; Semproni, S. P.; DeBeer, S.; Chirik, P. J. | |
113 | Bis(imino)pyridine Cobalt-Catalyzed Alkene Isomerization– Hydroboration: A Strategy for Remote Hydrofunctionalization with Terminal Selectivity.Obligacion, J. V.; Chirik, P. J. | |
112 | Cobalt Precursors for High-Throughput Discovery of Base Metal Asymmetric Alkene Hydrogenation Catalysts.Friedfeld, M. R.; Shevlin, M.; Hoyt, J. M.; Krska, S. W.; Tudge, M. T.; Chirik, P. J. | |
111 | Activation of Dinitrogen-Derived Hafnium Nitrides for Nucleophilic N-C Bond Formation with a Terminal Isocyanate.Semproni, S. P.; Chirik, P. J. | |
110 | Catalytic Hydrogenation Activity and Electronic Structure Determination of Bis(arylimidazol-2-ylidene)pyridine Cobalt Alkyl and Hydride Complexes.Yu, R. R.; Darmon, J. M.; Milsmann, C.; Margulieux, G. W.; Stieber, S. C. E.; DeBeer, S.; Chirik, P. J. | |
109 | Synthesis of a Base-Free Hafnium Nitride from N2 Cleavage: A Versatile Platform for Dinitrogen Functionalization.Semproni, S. P.; Chirik, P. J. | |
108 | Highly Selective Bis(imino)pyridine Iron-Catalyzed Alkene Hydroboration.Obligacion, J. V.; Chirik, P. J. | |
107 | Reversible Carbon- Carbon Bond Formation Induced by Oxidation and Reduction at a Redox-Active Cobalt Complex.Atienza, C. C. H.; Milsmann, C.; Semproni, S. P.; Turner, Z. R.; Chirik, P. J. | |
106 | Redox-Induced N2 Hapticity Switching in Zirconocene Dinitrogen Complexes.Semproni, S. P.; Knobloch, D. J.; Milsmann, C.; Chirik, P. J. | |
105 | Dinitrogen Borylation with Group 4 Metallocene Complexes.Semproni, S. P.; Chirik, P. J. | |
104 | Synthesis and electronic structure of bis(imino)pyridine iron metallocyclic intermediates in iron-catalyzed cyclization reactions.Hoyt, J. M.; Sylvester, K. T.; Semproni, S. P.; Chirik, P. J. | |
103 | Oxidation and reduction of bis(imino)pyridine iron dinitrogen complexes: Evidence for formation of a chelate trianion.Tondreau, A. M.; Stieber, S. C. E.; Milsmann, C.; Lobkovsky, E.; Weyhermüller, T.; Semproni, S. P.; Chirik, P. J. | |
102 | Oxidative addition of carbon-carbon bonds with a redox-active bis(imino)pyridine iron complex.Darmon, J. D.; Stieber, S. C. E.; Sylvester, K. T.; Fernández, I.; Lobkovsky, E.; Semproni, S. P.; Bill, E.; Wieghardt, K.; DeBeer, S.; Chirik, P. J. J. Am. Chem. Soc. 2012, 134, 17125-17137. AbstractAddition of biphenylene to the bis(imino)pyridine iron dinitrogen complexes, (iPrPDI)Fe(N2)2 and [(MePDI)Fe(N2)]2(μ2-N2) (RPDI = 2,6-(2,6-R2—C6H3—N═CMe)2C5H3N; R = Me, iPr), resulted in oxidative addition of a C—C bond at ambient temperature to yield the corresponding iron biphenyl compounds, (RPDI)Fe(biphenyl). The molecular structures of the resulting bis(imino)pyridine iron metallacycles were established by X-ray diffraction and revealed idealized square pyramidal geometries. The electronic structures of the compounds were studied by Mössbauer spectroscopy, NMR spectroscopy, magnetochemistry, and X-ray absorption and X-ray emission spectroscopies. The experimental data, in combination with broken-symmetry density functional theory calculations, established spin crossover (low to intermediate spin) ferric compounds antiferromagnetically coupled to bis(imino)pyridine radical anions. Thus, the overall oxidation reaction involves cooperative electron loss from both the iron center and the redox-active bis(imino)pyridine ligand. | 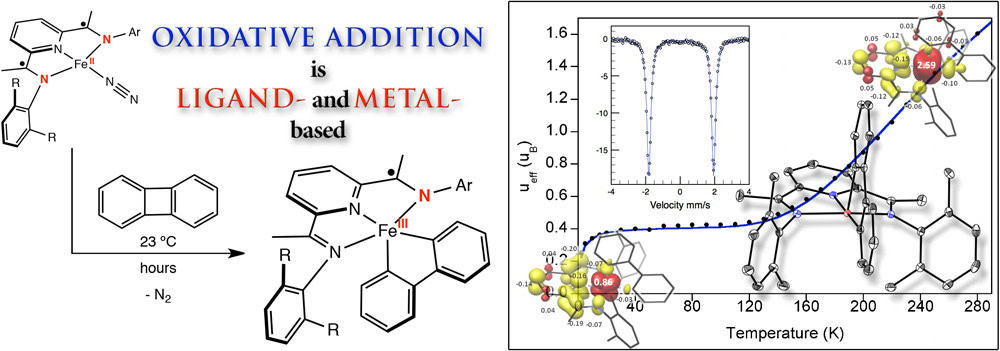 |
101 | High-selectivity bis(imino)pyridine iron catalysts for the hydrosilylation of 1,2,4-trivinylcyclohexane.Atienza, C. C. H.; Tondreau, A. M.; Weller, K. J.; Lewis, K. M.; Cruse, R. W.; Nye, S. A.; Boyer, J. L.; Delis, J. G. P.; Chirik, P. J. | |
100 | Di- and tetrametallic hafnocene oxamidides prepared from CO-induced N2 bond cleavage and thermal rearrangement to hafnocene cyanide derivatives.Semproni, S. P.; Margulieux, G. W.; Chirik, P. J. | |
99 | High-activity iron catalysts for the hydrogenation of hindered, unfunctionalized alkenes.Yu, R. P.; Darmon, J. M.; Hoyt, J. M.; Margulieux, G. W.; Turner, Z. R.; Chirik, P. J. | |
98 | Synthesis, electronic structure and alkene hydrosilylation activity of terpyridine and bis(imino)pyridine iron dialkyl complexes.Tondreau, A. M.; Atienza, C. C. H.; Darmon, J. M.; Milsmann, C.; Hoyt, H. M.; Weller, K. J.; Nye, S. A.; Lewis, K. M.; Boyer, J.; Delis, J. G. P.; Lobkovsky, E.; Chirik, P. J. | |
97 | Side-on dinitrogen complexes of titanocenes with disubstituted cyclopentadienyl ligands: Synthesis, structure and spectroscopic characterization.Semproni, S. P.; Milsmann, C.; Chirik, P. J. | |
96 | Structure and reactivity of a hafnocene μ-nitrido prepared from dinitrogen cleavage.Semproni, S. P.; Milsmann, C.; Chirik, P. J. | |
95 | Azo N=N bond cleavage with a redox- active vanadium compound involving metal-ligand cooperativity.Milsmann, C.; Turner, Z. R.; Semproni, S. P.; Chirik, P. J. | |
94 | Enantiopure, C1-symmetric bis(imino)pyridine cobalt complexes for asymmetric alkene hydrogenation.Monfette, S.; Turner, Z. R.; Semproni, S. P.; Chirik, P. J. J. Am. Chem. Soc. 2012, 134, 4561-4564. Highlighted in Nature Chemistry, 2012, 4, 337. | |
93 | Electronic effects in 4-substituted bis(imino)pyridines and the corresponding reduced iron compounds.Darmon, J. D.; Turner, Z. R.; Lobkovsky, E.; Chirik, P. J. | |
92 | Bis(imino)pyridine iron dinitrogen compounds revisited: Differences in electronic structure between four- and five-coordinate derivatives.Steiber, S. C. E.; Milsmann, C.; Hoyt, J. M.; Turner, Z. R.; Finkelstein, K. D.; Wieghardt, K.; DeBeer, S.; Chirik, P. J. | |
91 | Iron catalysts for selective anti-Markovnikov alkene hydrosilylation using tertiary silanes.Tondreau, A. M.; Atienza, C. C. H.; Weller, K. J.; Nye, S. A.; Lewis, K. M.; Delis, J. G. P.; Chirik, P. J. | |
90 | Studies into the mechanism of CO- induced N2 cleavage and C-C bond formation promoted by an ansa-hafnocene complex.Knobloch, D. J.; Semproni, S. P.; Lobkovsky, E.; Chirik, P. J. | |
89 | Synthesis and electronic structure determination of N-alkyl-substituted bis(imino)pyridine iron imides exhibiting spin cross behavior.Bowman, A. C.; Milsmann, C.; Bill, E.; Turner, Z. R.; Lobkovsky, E.; DeBeer, S.; Wieghardt, K.; Chirik, P. J. | |
88 | Preface: Forum on redox-active ligands.Chirik, P. J. | |
87 | Synthesis and electronic structure of reduced bis(imino)pyridine manganese compounds.Russell, S. K.; Bowman, A. C.; Lobkovsky, E.; Wieghardt, K.; Chirik, P. J. Eur. J. Inorg. Chem. 2011, 535-545. (Special issue on ligand-centered radicals). | |
86 | Dinitrogen silylation and cleavage with a hafnocene complex.Semproni, S. P.; Lobkovsky, E.; Chirik, P. J. | |
85 | Oxidation and reduction of bis(imino)pyridine iron dicarbonyl complexes.Tondreau, A. M.; Milsmann, C.; Lobkovsky, E.; Chirik, P. J. | |
84 | Synthesis, electronic structure and ethylene polymerization activity of bis(imino)pyridine cobalt alkyl cations.Atienza, C. C. H.; Milsmann, C.; Lobkovsky, E.; Chirik, P. J. Angew. Chem. Int. Ed. 2011, 50, 8143-8147 (Article Featured on Cover) | |
83 | An iron-catalyzed intermolecular [2π + 2π] cycloaddition.Russell, S. K.; Lobkovsky, E.; Chirik, P. J. | |
82 | Cyclisation of α, ω-dienes promoted by bis(indenyl)zirconium sandwich and ansa-titanocene dinitrogen complexes.Pun, D.; Knobloch, D. J.; Lobkovsky, E.; Chirik, P. J. Dalton Trans. 2011, 7737-7747 (Special Issue on d0 Metallocenes). | |
81 | Synthesis, electronic structure, and catalytic activity of reduced bis(aldimino)pyridine iron compounds: Experimental evidence for ligand participation.Russell, S. K.; Milsmann, C.; Lobkovsky, E.; Weyhermüller, T.; Chirik, P. J. | |
80 | Photolysis and thermolysis of bis(imino)pyridine cobalt azides: C-H activation from putative cobalt nitrido complexes.Atienza, C. C. H.; Bowman, A. C.; Lobkovsky, E.; Chirik, P. J. | |
79 | Functionalization of hafnium oxamidide complexes prepared from CO-induced N2 cleavage.Knobloch, D. J.; Lobkovsky, E.; Chirik, P. J. | |
78 | Synthesis and electronic structure of cationic, neutral, and anionic bis(imino)pyridine iron alkyl complexes: Evaluation of redox activity in single-component ethylene polymerization catalysts.Tondreau, A. M.; Milsmann, C.; Patrick, A. D.; Hoyt, H. M.; Lobkovsky, E.; Wieghardt, K.; Chirik, P. J. | |
77 | Carbon monoxide-induced dinitrogen cleavage with group 4 metallocenes: Reaction scope and coupling to N-H bond formation and C-O deoxygenation.Knobloch, D. J.; Lobkovsky, E.; Chirik, P. J. | |
76 | Reduced N-alkyl substituted bis(imino)pyridine cobalt complexes: Molecular and electronic structure for compounds varying by three oxidation states.Bowman, A. C.; Milsmann, C.; Bill, E.; Lobkovsky, E.; Weyhermüller, T.; Wieghardt, K.; Chirik, P. J. | |
75 | Cyclopentanone insertion into η9-indenyl rings of zirconium sandwich complexes.Pun, D.; Lobkovsky, E.; Keresztes, I.; Chirik, P. J. | |
74 | Group 4 transition metal sandwich complexes: Still fresh after almost 60 years.Chirik, P. J. | |
73 | Radical ligands confer nobility of base-metal catalysts.Chirik, P. J.; Wieghardt, K. W. | |
72 | Synthesis of aryl-substituted bis(imino)pyridine iron dinitrogen complexes.Russell, S. K.; Darmon, J. D.; Lobkovsky, E.; Chirik, P. J. | |
71 | Synthesis, molecular and electronic structures of reduced bis(imino)pyridine cobalt dinitrogen complexes: Ligand versus metal reduction.Bowman, A. C.; Milsmann, C.; Atienza, C. C. H. A.; Lobkovsky, E.; Wieghardt, K.; Chirik, P. J. | |
70 | Dinitrogen cleavage and functionalization by carbon monoxide promoted by a hafnium complex.Knobloch, D. J.; Lobkovsky, E.; Chirik, P. J. | |
69 | Addition of methyl triflate to a hafnocene dinitrogen complex: Stepwise N2 methylation and conversion to a hafnocene hydrazonato compound.Knobloch, D. J.; Benito-Garagorri, D.; Bernskoetter, W. H.; Keresztes, I.; Lobkovsky, E.; Toomey, H.; Chirik, P. J. | |
68 | Nitrogen fixation – one electron at a time.Chirik, P. J. | |
67 | 1,4-Addition of alkyl halides to a side-on bound hafnocene dinitrogen complex.Benito-Garagorri, D.; Bernskoetter, W. H.; Lobkovsky, E.; Chirik, P. J. | |
66 | Dinitrogen complexes of bis(cyclopentadienyl) titanium derivatives: Structure diversity arising from substituent manipulation.Hanna, T. E; Lobkovsky, E.; Chirik, P. J. | |
65 | Enantiopure pyridine bis(oxazoline) ‘Pybox’ and bis(oxazoline) ‘Box’ iron dialkyl complexes: Comparison to bis(imino)pyridine compounds and application to catalytic hydrosilylation of ketones.Tondreau, A. M.; Darmon, J. M.; Wile, B. M.; Floyd, S. K.; Lobkovsky, E.; Chirik, P. J. | |
64 | Iron-catalyzed, hydrogen-mediated reductive cyclization of 1,6- enynes and diynes: Evidence for bis(imino)pyridine ligand participation.Sylvester, K. T.; Chirik, P. J. | |
63 | Synthesis of bis(imino)pyridine iron amide and ammonia compounds from an N-H transfer agent.Bowman, A. C.; Bart, S. C.; Heinemann, F. W.; Meyer, K.; Chirik, P. J. | |
62 | Bis(indenyl)hafnium chemistry: Ligand-induced haptotropic rearrangement and fundamental reactivity studies at a reduced hafnium center.Pun, D.; Leopold, S. M.; Bradley, C. A.; Lobkovsky, E.; Chirik, P. J. | |
61 | Iron Catalysis in Organic Chemistry: Reactions and Applications.Chirik, P. J. | |
60 | N-N bond cleavage in diazoalkanes by a bis(imino)pyridine iron complex.Russell, S. K.; Lobkovsky, E.; Chirik, P. J. | |
59 | Reduction chemistry of aryl- and alkyl-substituted bis(imino)pyridine iron dihalide compounds: Molecular and electronic structures of [(PDI)2Fe] derivatives.Wile, B. M.; Trovitch, R. J.; Bart, S. C.; Tondreau, A. M.; Lobkovsky, E.; Milsmann, C.; Bill, E.; Wieghardt, K.; Chirik, P. J. | |
58 | Carbon-oxygen bond cleavage in bis(imino)pyridine compounds: Catalyst deactivation pathways and observation of acyl C-O bond cleavage in esters.Trovitch, R.; Lobkovsky, E.; Bouwkamp, M.; Chirik, P. J. | |
57 | N2 hydrogenation from activated end-on bis(indenyl)zirconium dinitrogen complexes.Pun, D.; Bradley, C. A.; Lobkovsky, E.; Keresztes, I.; Chirik, P. J. | |
56 | Bis(imino)pyridine iron alkyls containing β-hydrogens: Synthesis, evaluation of kinetic stability, and decomposition pathways involving chelate participation.Trovitch, R. J.; Lobkovsky, E.; Chirik, P. J. | |
55 | Bis(imino)pyridine iron complexes for aldehyde and ketone hydrosilylation.Tondreau, A. M.; Lobkovsky, E.; Chirik, P. J. | |
54 | Indenyl dinitrogen chemistry: N2 coordination to an isolated zirconium sandwich and synthesis of side-on, end-on dinitrogen compounds.Pun, D.; Lobkovsky, E.; Chirik, P. J. | |
53 | Carboxylation of an ansa-zirconocene dinitrogen complex: Regiospecific hydrazine synthesis from N2 and CO2.Knobloch, D. J.; Toomey, H. E.; Chirik, P. J. | |
52 | Functional group tolerance and substrate scope in bis(imino)pyridine iron catalyzed alkene hydrogenation.Trovitch, R. J.; Lobkovsky, E.; Bill, E.; Chirik, P. J. | |
51 | 1,2-Addition versus σ-Bond metathesis reactions in transient bis(cyclopentadienyl)zirconium imidos: Evidence for a d0 dihydrogen complex.Toomey, H. E.; Pun, D.; Veiros, L. F.; Chirik, P. J. | |
50 | Synthesis of bis(imino)pyridine iron di- and monoalkyl complexes: Stability differences between FeCH2SiMe3 and FeCH2CMe3 derivatives.Fernández, I.; Trovitch, R. J.; Lobkovsky, E.; Chirik, P. J. | |
49 | Neutral-ligand complexes of bis(imino)pyridine iron: Synthesis, structure and spectroscopy.Bart, S. C.; Lobkovsky, E.; Bill, E.; Weighardt, K.; Chirik, P. J. | |
48 | Amineborane dehydrogenation promoted by isolable zirconium sandwich, titanium sandwich and N2 complexes.Pun, D.; Lobkovsky, E.; Chirik, P. J. | |
47 | Iron diazoalkane chemistry: N-N bond hydrogenation and intramolecular C-H activation.Bart, S. C.; Bowman, A. C.; Lobkovsky, E.; Chirik, P. J. | |
46 | C-O and C-S bond cleavage in chelating diethers and thioethers promoted by η9, η5-bis(indenyl)zirconium sandwich complexes: A combined experimental and computational study.Bradley, C. A.; Veiros, L. F.; Chirik, P. J. | |
45 | Bis(cyclopentadienyl)titanium dinitrogen chemistry: Synthesis and characterization of a side-on bound haptomer.Hanna, T. E.; Bernskoetter, W. H.; Bouwkamp, M. W.; Lobkovsky, E.; Chirik, P. J. | |
44 | Dihydrogen and silane addition to base-free, monomeric bis(cyclopentadienyl) titanium oxides.Hanna, T. E.; Lobkovsky, E.; Chirik, P. J. | |
43 | N-H group transfer and oxidative addition chemistry promoted by isolable bis(cyclopentadienyl)titanium sandwich complexes.Hanna, T. E.; Lobkovsky, E.; Chirik, P. J. | |
42 | Diazene dehydrogenation follows H2 addition to coordinated dinitrogen in an ansa-zirconocene complex.Hanna, T. E.; Keresztes, I.; Lobkovsky, E.; Chirik, P. J. | |
41 | Nitrogen-carbon bond formation from N2 and CO2 promoted by a hafnocene dinitrogen complex yields a substituted hydrazine.Bernskoetter, W. H.; Lobkovsky, E.; Chirik, P. J. Angew. Chem. Int. Ed. 2007, 2858-2861. (Article Featured on Cover) | |
40 | Dinitrogen functionalization with bis(cyclopentadienyl) complexes of zirconium and hafnium.Chirik, P. J. Dalton Trans. 2007, 16-26. (Invited Feature Article, Featured on Cover). | |
39 | Carbon-oxygen bond cleavage with η9, η5-bis(indenyl)zirconium sandwich complexes.Bradley, C. A.; Veiros, L. F.; Pun, D.; Lobkovsky, E.; Keresztes, I. Chirik, P. J. | |
38 | An Fe(VI) nitride: There is plenty of room at the top!Chirik, P. J. | |
37 | Electronic structure of bis(imino)pyridine iron dichloride, monochloride and neutral ligand complexes: A combined, structural, spectroscopic and computational study.Bart, S. C.; Chlopek, K.; Bill, E.; Bouwkamp, M. W.; Lobkovsky, E.; Neese, F.; Wieghardt, K.; Chirik, P. J. | |
36 | Iron-catalyzed [2π + 2π] cycloaddition of α, ω-dienes: The importance of redox-active supporting ligands.Bouwkamp, M. W.; Bowman, A. C. Lokbovsky, E.; Chirik, P. J. | |
35 | Bis(diisopropylphosphino)pyridine iron dicarbonyl, dihydride, and silyl hydride complexes.Trovitch, R. J.; Lobkovsky, E.; Chirik, P. J. | |
34 | Arene coordination in bis(imino)pyridine iron complexes: Identification of catalyst deactivation pathways in iron- catalyzed hydrogenation and hydrosilation.Archer, A. M.; Bouwkamp, M. W.; Cortez, M. –P.; Lobkovsky, E.; Chirik, P. J. | |
33 | N-C bond formation promoted by a hafnocene dinitrogen complex: Comparison of zirconium and hafnium congeners.Bernskoetter, W. H.; Olmos, A. V.; Pool, J. A.; Lobkovsky, E.; Chirik, P. J. | |
32 | Synthesis of bis(indenyl)zirconium dihydrides and subsequent rearrangement to η5, η3-4,5-dihydroindendiyl ligands: Evidence for intermediates during the hydrogenation to tetrahydroindenyl derivativesBradley, C. A.; Lobkovsky, E.; Keresztes, I.; Chirik, P. J. | |
31 | Mono(dinitrogen) and carbon monoxide adducts of bis(cyclopentadienyl) titanium sandwichesHanna, T. E.; Lobkovsky, E.; Chirik, P. J. | |
30 | Synthesis and hydrogenation of bis(imino)pyridine iron imidesBart, S. C.; Lobkovsky, E.; Bill, E.; Chirik, P. J. | |
29 | The ‘Indenyl Effect’ in zirconocene dihydride chemistry.Bradley, C. A.; Keresztes, I.; Lobkovsky, E.; Chirik, P. J. | |
28 | Carbon-hydrogen bond activation with a cyclometalated zirconocene hydride: Mechanistic differences between arene and alkane reductive elimination.Bernskoetter, W. H.; Pool, J. A.; Lobkovsky, E.; Chirik, P. J. | |
27 | N2 hydrogenation promoted by a side-on bound hafnocene dinitrogen complex.Bernskoetter, W. H.; Olmos, A. V.; Lobkovsky, E.; Chirik, P. J. | |
26 | Bis(imino)pyridine ligand deprotonation promoted by a transient iron amideBouwkamp, M. W.; Lobkovsky, E.; Chirik, P. J. Inorg. Chem. 2006, 45, 2-4. (Article featured on the cover). | |
25 | Ancillary ligand effects on C-H bond activation reactions promoted by β-diiminate iridium complexes.Bernskoetter, W. H.; Lobkovsky, E.; Chirik, P. J. | |
24 | Low-valent α-diimine complexes for catalytic olefin hydrogenation.Bart, S. C.; Hawrelak, E. J.; Lobkovsky, E.; Chirik, P. J. | 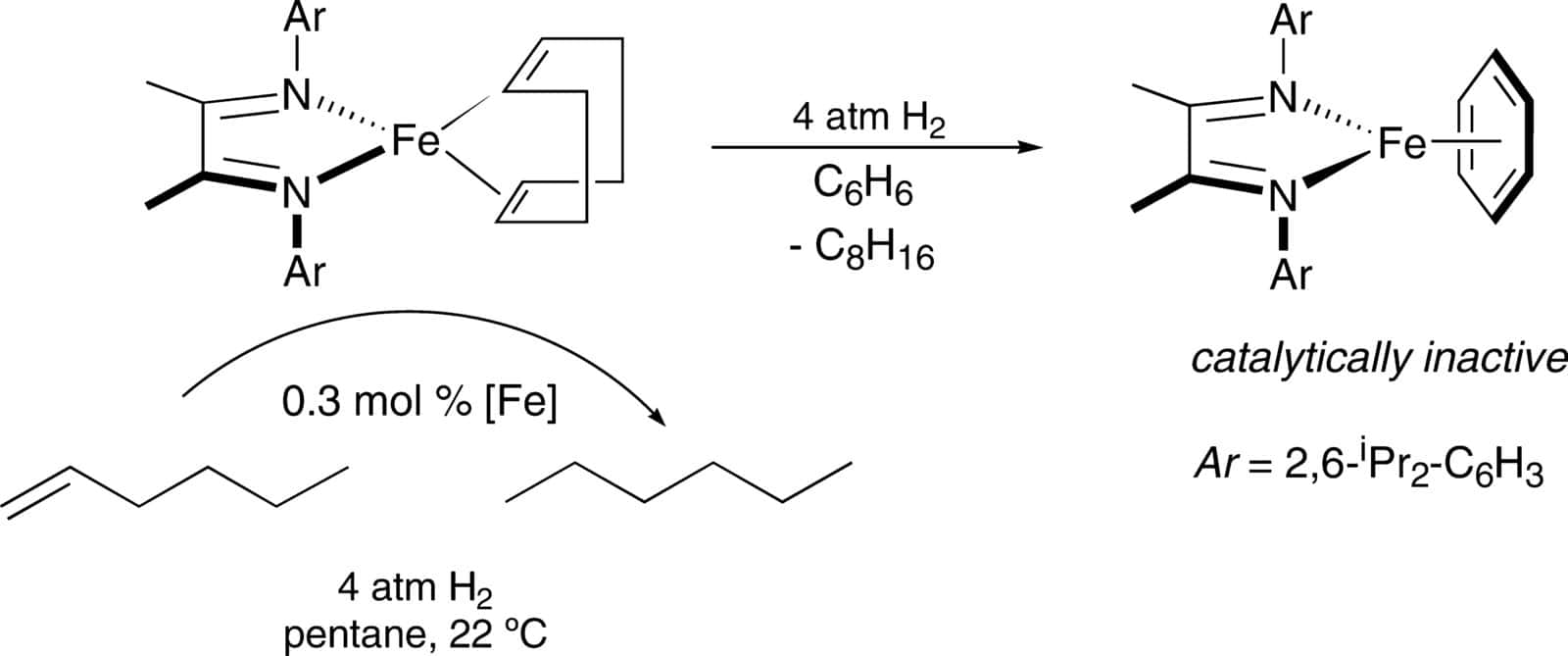 |
23 | Kinetics and mechanism of N2 hydrogenation in bis(cyclopentadienyl)zirconium complexes and dinitrogen functionalization by 1,2-addition of a saturated C-H bond.Bernskoetter, W. H.; Lobkovsky, E.; Chirik, P. J. | |
22 | C-H bond activation reactions with ligand adducts of a β-diiminate iridium dihydride.Bernskoetter, W. H.; Lobkovsky, E.; Chirik, P. J. | |
21 | Ligand-induced haptotropic rearrangements in bis(indenyl)zirconium sandwich complexes.Bradley, C. A.; Lobkovsky, E.; Keresztes, I.; Chirik, P. J. | |
20 | Bis(imino)pyridine iron(II) alkyl cations for olefin polymerization.Bouwkamp, M. W.; Lobkovsky, E.; Chirik, P. J. | |
19 | Square planar bis(imino)pyridine iron halide and alkyl complexes.Bouwkamp, M. W.; Bart, S. C.; Hawrelak, E. J.; Trovitch, R. J.; Lobkovsky, E.; Chirik, P. J. | |
18 | Dinitrogen functionalization with terminal alkynes, amines and hydrazines promoted by [(η5-C5Me4H)2Zr]2(μ2, η2, η2-N2): Observation of side-on and end-on diazenido complexes in the reduction of N2 to hydrazine.Bernskoetter, W. H.; Pool, J. A.; Lobkovsky, E.; Chirik, P. J. | |
17 | Square planar versus tetrahedral geometry in four coordinate iron(II) complexes.Hawrelak, E. J.; Bernskoetter, W. H.; Lobkovsky, E.; Yee, G.; Bill, E.; Chirik, P. J. | 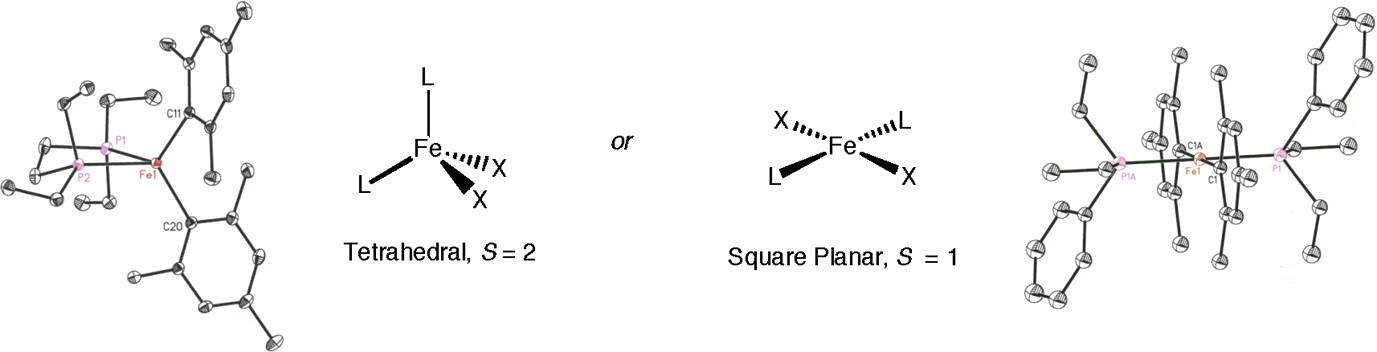 |
16 | The importance of cyclopentadienyl substituent effects in group 4 metallocene dinitrogen chemistry.Pool, J. A.; Chirik, P. J. | |
15 | Zirconium sandwich complexes with η9 indenyl ligands: Well-defined precursors for zirconocene-mediated coupling reactions.Bradley, C. A.; Keresztes, I.; Lobkovsky, E.; Young, V. G.; Chirik, P. J. | |
14 | Dinitrogen activation by titanium sandwich complexes.Hanna, T. E.; Lobkovsky, E.; Chirik, P. J. | |
13 | On the origin of dinitrogen hydrogenation promoted by [(η5-C5Me4H)2Zr]2(μ2, η2, η2-N2).Pool, J. A.; Bernskoetter, W. H.; Chirik, P. J. | |
12 | Synthesis and characterization of zirconium and iron complexes containing substituted indenyl ligands: Evaluation of steric and electronic parameters.Bradley, C. A.; Flores-Torres, S.; Lobkovsky, E.; Abruna, H. D.; Chirik, P. J. | |
11 | Preparation and molecular and electronic structures of iron(0) dinitrogen and silane complexes and their application to catalytic hydrogenation and hydrosilation.Bart, S. C.; Lobkovsky, E.; Chirik, P. J. | |
10 | Synthesis of a β-diiminate iridium tetrahydride for arene C-H bond activation.Bernskoetter, W. H.; Lobkovsky, E.; Chirik, P. J. | |
9 | Hydrogenation and cleavage of dinitrogen to ammonia with a well-defined zirconium complex.Pool, J. A.; Lobkovsky, E.; Chirik, P. J. | |
8 | Synthesis of a base- free titanium imido and a transient alkylidene from a titanocene dinitrogen complex. Studies on Ti=NR hydrogenation, nitrene group transfer and comparison of 1,2-addition rates.Hanna, T. E.; Keresztes, I.; Lobkovsky, E.; Bernskoetter, W. H., Chirik, P. J. | |
7 | Synthesis, reactivity and solid state structures of four-coordinate iron(II) and manganese(II) alkyl complexes.Bart, S. C.; Hawrelak, E. J.; Schmisseur, A. K.; Lobkovsky, E.; Chirik, P. J. | |
6 | Synthesis of a zirconium sandwich complex and crystallographic characterization of its adduct with tetrahydrofuran.Bradley, C. A.; Lobkovsky, E.; Chirik, P. J. | |
5 | Alkyl substituent effects on reductive elimination reactions in zirconocene alkyl hydride complexes. Manipulation of the alkyl steric environment allows the synthesis of a zirconocene dinitrogen complex.Pool, J. A.; Lobkovsky, E.; Chirik, P. J. | |
4 | Cyclopentadienyl substituent effects on reductive elimination reactions in group 4 metallocenes: kinetics, mechanism and application to dinitrogen activation.Pool, J. A.; Lobkovsky, E.; Chirik, P. J. | |
3 | Selective, catalytic carbon-carbon bond activation and functionalization promoted by late transition metal catalysts.Bart, S. C.; Chirik, P. J. | |
2 | Functionalization of elemental phosphorus with [(η5- C5Me5)(η5-C5H5tBu)ZrH2]2.Pool, J. A.; Lobkovsky, E.; Chirik, P. J. | |
1 | A convenient method for the preparation of zirconocene hydrido chloride, isobutyl hydride and dihydride complexes with tert-butyl lithium.Pool, J. A.; Bradley, C. A.; Chirik, P. J. | |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3))-C4H5N]). DMCOD was isolated in 92% yield, at the preparative scale, with a catalyst loading of 0.025 mol%, and a TON of 3680. Catalytic hydrogenation of DMCOD yielded 1,4-dimethylcyclooctane (DMCO). The cyclic structure and ring strain of DMCO afforded gravimetric and volumetric net heats of combustion 2.4 and 9.2% higher, respectively, than conventional jet fuel. In addition, the presence of methyl branches at two sites resulted in a −20 °C kinematic viscosity of 4.17 mm2 s−1, 48% lower than the maximum allowed value for conventional jet fuel. The ability to derive isoprene and related alcohols readily from abundant biomass sources, coupled with the highly efficient [Fe]-catalyzed [4 + 4]-cycloaddition described herein, suggests that this process holds great promise for the economical production of high-performance, bio-based jet fuel blendstocks.
C(CH3))-C4H5N]). DMCOD was isolated in 92% yield, at the preparative scale, with a catalyst loading of 0.025 mol%, and a TON of 3680. Catalytic hydrogenation of DMCOD yielded 1,4-dimethylcyclooctane (DMCO). The cyclic structure and ring strain of DMCO afforded gravimetric and volumetric net heats of combustion 2.4 and 9.2% higher, respectively, than conventional jet fuel. In addition, the presence of methyl branches at two sites resulted in a −20 °C kinematic viscosity of 4.17 mm2 s−1, 48% lower than the maximum allowed value for conventional jet fuel. The ability to derive isoprene and related alcohols readily from abundant biomass sources, coupled with the highly efficient [Fe]-catalyzed [4 + 4]-cycloaddition described herein, suggests that this process holds great promise for the economical production of high-performance, bio-based jet fuel blendstocks.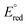 = −1.32 V vs. Fc/Fc+) with the most acidic N–H bond (pKa < 2.6, THF), whereas the ligands CH3CN (
= −1.32 V vs. Fc/Fc+) with the most acidic N–H bond (pKa < 2.6, THF), whereas the ligands CH3CN (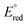 = −1.60 V, pKa < 5.5) and I− (
= −1.60 V, pKa < 5.5) and I− (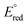 = −2.03 V, pKa = 9.3) gave more reducing complexes with less acidic N–H bonds. Computational (DFT) studies confirm weak N–H bond strengths of 32.8 (L = I−), 35.4 (L = CH3CN) and 36.2 kcal mol−1 (L = 3,5-(CF3)2C6H3CN) in the hydrazido series.
= −2.03 V, pKa = 9.3) gave more reducing complexes with less acidic N–H bonds. Computational (DFT) studies confirm weak N–H bond strengths of 32.8 (L = I−), 35.4 (L = CH3CN) and 36.2 kcal mol−1 (L = 3,5-(CF3)2C6H3CN) in the hydrazido series.